
Here are some highlights, from Ivermectin studies, that show why it works so well. These highlighted excerpts below are copied and pasted from the studies further down the page. So please note that these cut and pasted highlighted excerpts have been pieced together from the studies further down the page, and may not appear to be in their proper context.
Pierre Kory also explains why it works well, in the videos on THIS PAGE about Ivermectin's efficacy.
A coronavirus has 4 basic proteins. the spike protein (S), the envelope protein (E), the nucleocapsid protein (N) and the membrane protein (M). The N protein is incorporated in the RNA helical structure, which underlies the envelope (15). Overall, the N protein enhances coronavirus transcription, interacts with the viral genome and with M in the viral envelope. Inhibition of N was shown to greatly suppress viral replication, suggesting it is an essential factor in efficient virion production (14, 15).
N is the highest expressed protein in infected cells, further corroborating its importance in the viral life cycle (15).
N is proposed to dampen the host cell’s antiviral transcriptional response within the nucleus (8).
The importance of blocking the nuclear import of viral proteins emerged when it was later shown that IVM also prevented replication of HIV (36)
Fifty years later, this same drug is suddenly at the forefront of the race against the current pandemic, namely via its unintentional inhibition of nuclear transport.
Its antiparasitic effects are primarily caused by high-affinity irreversible binding to glutamate-gated chloride (Cl-) channels located on nerve and muscle cells of nematode, which leads to hyperpolarization (9, 31). Ultimately, the increased permeability to Cl- results in paralysis and death of the nematode (31).
severe detrimental effects in humans were shown only in those who over-dosed using approximately 15.4 mg/kg body weight IVM, which is 77 times above the prescribed dose.
The Wagstaff et al., 2011 and 2012 studies were pivotal in providing much of the initial rationale for the recent consideration of IVM as a SARS-CoV-2 antiviral agent. In fact, it was the same researchers who nine years later, in 2020, demonstrated that IVM inhibits SARS-CoV-2 in vitro replication (8).
Given its efficacy in inhibiting nuclear import of other viral proteins, the anti-viral effect of IVM against SARS-CoV-2 was evaluated shortly after the pandemic erupted (8). Specifically, Vero/hSLAM cells were inoculated with SARS-CoV-2 isolate for 2 hrs., followed up with supplementation of 5 μM of IVM. Within 24 hrs. after treatment, there was a 93% reduction of viral RNA in the supernatant and 99.8% reduction of cellular viral RNA, compared to controls. After 48 hrs., there was a further 5000-fold reduction of viral RNA in the supernatant as well as the cell pellets, indicating that cells were essentially ‘cleared’ of SARS-CoV-2 (8). Although IVM possessed a potent antiviral activity (IC50= ~2μM), no cytotoxicity was detected at any time points in this study (8).
IVM Specifically Interacts With IMPα
IVM was shown to specifically inhibit IMP α/β mediated nuclear import required for replication of HIV-1 and Dengue virus, and therefore it was proposed as the potential mechanism by which it inhibits SARS-CoV-2 (36). Indeed, this baton was passed on, and a subsequent study verified the IVM-IMPα interaction in host cells (38). In fact, it was shown that IVM not only inhibits IMPα association with IMPβ, but can even dissociate IMPα/β heterodimers (38). The specific binding target of IVM was identified, using CD spectroscopy, to be the alpha-helical rich ‘armadillo’ (ARM) domain of IMPα. Moreover, as concentrations of IVM increased, alpha helices in the ARM domain became increasingly destabilized. No changes were detected in the structure of IMPβ. They further verified that this observed effect on IMPα impaired its binding to NLS-containing nsp5 from Dengue Virus (38). As such, preventing N interaction with IMPα, is a likely mechanism that contributes to IVM’s ability to hinder SARS-CoV-2 in vitro replication (Figure 2).
Because the nucleocapsid (N) protein contains a nuclear localization signal, IVM is expected to prevent the binding of IMPα to the N binding site. Consequently, N would not perform its nuclear activity which is thought to suppress the host immune response and sequester ribosomal subunits, mechanisms which are thought to abrogate sufficient viral replication. In addition, the expression of two major cytokines, TNFα and IL-6 which drive the detrimental cytokine storm in COVID-19 patients were also shown to be dampened in the presence of IVM. As of yet, these two major mechanisms which involve viral replication and immune response suppression appear to characterize the main activities of IVM against SARS-CoV-2.
Here are the studies, from which the above highlights were taken. To see the original studies, just click on the links pasted above the studies.
https://www.frontiersin.org/articles/10.3389/fimmu.2021.663586/full
Repurposing Ivermectin for COVID-19: Molecular Aspects and Therapeutic Possibilities
 Zena Wehbe1†,
Zena Wehbe1†,  Maya Wehbe2†,
Maya Wehbe2†,  Rabah Iratni3,
Rabah Iratni3,  Gianfranco Pintus4,5,
Gianfranco Pintus4,5,  Hassan Zaraket6,7,
Hassan Zaraket6,7,  Hadi M. Yassine8* and
Hadi M. Yassine8* and  Ali H. Eid9,10*
Ali H. Eid9,10*- 1Department of Biology, Faculty of Arts and Sciences, American University of Beirut, Beirut, Lebanon
- 2Department of Internal Medicine, Basingstoke & North Hampshire Hospital, Basingstoke, United Kingdom
- 3Department of Biology, College of Science, United Arab Emirates University, Al-Ain, United Arab Emirates
- 4Department of Medical Laboratory Sciences, College of Health Sciences, and Sharjah Institute for Medical Research, University of Sharjah, Sharjah, United Arab Emirates
- 5Department of Biomedical Sciences, University of Sassari, Sassari, Italy
- 6Department of Experimental Pathology, Immunology and Microbiology, Faculty of Medicine, American University of Beirut, Beirut, Lebanon
- 7Center for Infectious Disease Research (CIDR), Faculty of Medicine, American University of Beirut, Beirut, Lebanon
- 8Biomedical Research Center, Q.U. Health, Qatar University, Doha, Qatar
- 9Department of Basic Medical Sciences, College of Medicine, Q.U. Health. Qatar University, Doha, Qatar
- 10Biomedical and Pharmaceutical Research Unit, Q.U. Health, Qatar University, Doha, Qatar
As of January 2021, SARS-CoV-2 has killed over 2 million individuals across the world. As such, there is an urgent need for vaccines and therapeutics to reduce the burden of COVID-19. Several vaccines, including mRNA, vector-based vaccines, and inactivated vaccines, have been approved for emergency use in various countries. However, the slow roll-out of vaccines and insufficient global supply remains a challenge to turn the tide of the pandemic. Moreover, vaccines are important tools for preventing the disease but therapeutic tools to treat patients are also needed. As such, since the beginning of the pandemic, repurposed FDA-approved drugs have been sought as potential therapeutic options for COVID-19 due to their known safety profiles and potential anti-viral effects. One of these drugs is ivermectin (IVM), an antiparasitic drug created in the 1970s. IVM later exerted antiviral activity against various viruses including SARS-CoV-2. In this review, we delineate the story of how this antiparasitic drug was eventually identified as a potential treatment option for COVID-19. We review SARS-CoV-2 lifecycle, the role of the nucleocapsid protein, the turning points in past research that provided initial ‘hints’ for IVM’s antiviral activity and its molecular mechanism of action- and finally, we culminate with the current clinical findings.
Introduction
SARS-CoV-2 is a positive-sense RNA β-coronavirus, enclosing a capped polyadenylated 30 kb genome, which is the largest among RNA viruses (1). SARS-CoV-2 binds to the ACE2 enzyme on the surface of the target host cell by way of its outer spike protein (S) (2). The receptor-binding domain (RBD) on the S1 subunit interacts with the peptidase domain of ACE2. After partitioning into the host membrane, sequential enzymatic cleavages ultimately lead to the release of the viral genome into the cell (3).
The development of successful vaccines has been a priority in the pharmaceutical and scientific community (4). However, the time between the initial SARS-CoV-2 outbreak in December 2019 until the pharmaceutical companies began vaccine distribution spanned over a year (5). During this period, two million people have died worldwide, according to the World Health Organization (WHO). Moreover, the increasing mutations detected in the S protein have raised concerns that virus evolution might outpace vaccine rollout and the time needed to reach herd immunity (6, 7). Additionally, while vaccines are the main stay for halting the pandemic, it remains critical to develop therapeutics to treat patients and reduce the disease burden.
The drug ivermectin (IVM) has recently been shown to inhibit replication of SARS-CoV-2 in cell cultures (8). IVM is a widely used drug, known best for its antiparasitic properties in both veterinary and human medicine. It was first discovered in the 1970s by microbiologist Satoshi Omura and parasitologist William Campbell (9). Fifty years later, this same drug is suddenly at the forefront of the race against the current pandemic, namely via its unintentional inhibition of nuclear transport. It is important to understand and elucidate the ‘journey’ of how IVM emerged as a therapeutic agent against SARS-CoV-2, to follow this precedent and encourage repurposing available drugs for an increasing number of diseases. As such, we aim to highlight essential steps and components in the SARS-CoV-2 lifecycle, the significance of the nucleocapsid protein, the anecdotal evidence that hinted its potential as an anti-viral drug and its molecular mechanism of action. Finally, we summarize real-time results of current clinical trials.
SARS-CoV-2 Lifecycle
Initial Formation of the Replicase-Transcriptase Complexes
The basis of the seemingly successful repurposing of IVM is rooted in the identification of important components encoded by the viral genome. The SARS-CoV-2 viral genome encodes non-structural, structural, and accessory proteins. Its positive mRNA strand is translated within the host cell in order to, first, produce its own replication machinery, and second, to produce the structural components required to house viral progeny (10). Two-thirds of the genome code for two large polyproteins, pp1a and pp1ab. Once formed, the polyproteins are subsequently cleaved into 16 individual non-structural proteins (nsps), which primarily provide enzymatic activity (11). Three nsps (1–3) are cleaved by papain-like proteases (PLpro), which itself is localized within nsp3, and the rest are cleaved by the main protease (3C-like protease, 3CLpro) on nsp5 (1). As such, translation of the viral PLpro and 3CLpro are essential for efficient reproduction of the virus. Once the nsps are available, they cooperatively form the replicase-transcriptase complexes (RTCs), which are required for the production of new virions (12). Some nsps (3,4 and 6) induce the development of double membranes from the endoplasmic reticulum (E.R.), Golgi apparatus (G.A.) or the ER-Golgi intermediate compartment (ERGIC), which serve as foci for viral genesis (12). Collectively, the rest of the nsps in the RTC include RNA polymerase, helicase, exoribonuclease, and methyltransferase, among many others. The exact mechanism of replicating its own genome is still under investigation. However, it is understood that negative-sense intermediates are initially formed and then serve as templates for reproducing both genomic and sub-genomic positive-sense RNAs (13). A potential model for the RNA replication in SARS-CoV-2 has been postulated and it is based on homologous proteins in SARS-CoV-1 (10).
The Importance of the Nucleocapsid Protein
Structural proteins are highly conserved among the various genera of coronaviruses. They include the spike protein (S), the envelope protein (E), the nucleocapsid protein (N) and the membrane protein (M).
(pic taken from the web)

Once the structural proteins are synthesized, and the viral RNA is reproduced, the S, M and E become embedded within the previously formed double membranes from the host E.R. and eventually reach ERGIC. Meanwhile, the N protein which is tethered to the newly formed genome ‘delivers’ this RNA into S-M-E-embedded ERGIC membrane, leading to the formation of ‘pockets’ which eventually seal off into new virions (1). The interaction of N with the 3’-end of the viral genome is mediated via nsp3 (14), the largest subunit of the RTC. The nsp3 acidic ubiquitin-like N terminal domain (UbI1) binds to a serine- and arginine-rich domain in the N protein, thereby anchoring the viral genome to the RTC in order to facilitate RNA replication and, importantly, to eventually ensure the localization of the newly synthesized genome within the viral envelope (Hurst, Koetzner, & Masters, 2013). Ultimately, the N protein is incorporated in the RNA helical structure, which underlies the envelope (15). Overall, the N protein enhances coronavirus transcription, interacts with the viral genome and with M in the viral envelope. Notably, inhibition of N was shown to greatly suppress viral replication, suggesting it is an essential factor in efficient virion production (14, 15). Interestingly, N is the highest expressed protein in infected cells, further corroborating its importance in the viral life cycle (15).
The SARS-CoV-2 Nucleocapsid Protein Enters the Nucleus
The Role of Importins
Although RNA replication and translation occur in the cytosol, nuclear access is a key event in the infectious cycle of several viruses, including coronaviruses (1, 8). However, the entry of proteins into the nucleus is a tightly regulated process. To evade this limiting barrier, some viral proteins exploit the importin (IMP) superfamily of nuclear transporters to gain nuclear access (16). Nucleocytoplasmic trafficking is mediated via transmembrane nuclear pore complexes (NPCs) in the nucleus, composed of nucleoporin (NPR) subunits. A major class of NPRs known as FG-NPRs are distributed throughout the NPCs and enable nucleocytoplasmic transport due to their interaction with IMP transporters (17). The major IMP classes include IMPα and IMPβ. Nuclear import is mainly mediated either by IMPβs or by heterodimers of IMPα/IMPβ1 (17–19). For cytosolic protein cargo destined for nuclear import, IMPs, particularly IMPα proteins, recognize nuclear localization signals (NLS) on target cargo proteins, whereas IMPβ facilitates the actual transport via the NPCs (18). Efficient target binding to the IMPs/EXPs is further supported by Ran, the small monomeric GTPase (20). Active Ran causes dissociation of IMPβ from the importin/NLS-protein complex, releasing its tethered cargo into the nucleus (21). Thus, for a potential SARS-CoV-2 protein to reach the nucleus, it must contain an NLS, properly interact with IMP proteins and Ran must be activated.
SARS-CoV-2 Nucleocapsid Protein Contains an Enhanced Nuclear Localization Signal
As it happens, the SARS-CoV-2 N contains NLS motifs. Of great significance is the finding that NLS regions in the N gene of SARS-CoV viruses are highly variable compared to the NLS of other coronavirus clades (22). Importantly, these changes occurred during the recent evolution of the highly pathogenic coronavirus clades- including SARS-CoV-2 (22). Incidentally, the numerous nucleotide insertions and deletions within the NLS are associated with enhanced nuclear translocation. Three NLS motifs have been detected on the N of SARS-CoV-2, SARS-CoV, MERS-CoV and seasonal coronaviruses. Uniquely, as a result of the nucleotide variations found in SARS-CoV-2 and SARS-CoV-1, all three NLS motifs contain a distinctly higher overall positive charge among the peptides compared to the less virulent coronaviruses. The higher positive charge of NLS renders the entire N protein also more positively charged and subsequently enhances its efficacy (23). It has been previously corroborated in animal studies that the enhanced translocation of viral Ns to the nucleus results in more severe pathogenicity (24). Therefore, it is possible that these more positively charged Ns, which are characteristic of SARS-CoV-2, may be partially responsible for the associated detrimental effects.
The Putative Role of the Nucleocapsid Protein Within the Nucleus
It was previously shown that viral proteins that enter the nucleus might suppress host genes related to the anti-viral response, leading ultimately to increased pathogenicity (25). This may also be the case with SARS-CoV-2, as in vitro studies indicated that the SARS-CoV-2 NP could interact with dsDNA, possibly due to its high positive charge and the negative charge of DNA (26). Although the exact activity of the SARS-CoV-2 N within the nucleus has not been fully characterized, previous examination of several coronavirus Ns can offer insight (24).
The N of the coronavirus infectious bronchitis virus (IBV) was detected not only in the cytoplasm but also within the nucleolus. Nucleolus targeting was also shown with the SARS-CoV-1 N (27). It is important to note that the presence of N in the nucleus was indispensable for the replication of IBV, highlighting that cytosolic activity was not sufficient. In another related coronavirus, mouse hepatitis virus (MHV), nuclear proteins were also implicated in its replication. MHV N was specifically detected in the nucleolus, which itself is formed during interphase of the cell cycle and allows formation of ribosomal RNA (rRNA) and ribosomal subunits. The reason for N targeting of the nucleolus is not entirely understood. However, it is possible that N associates with rRNAs, in order to ‘reserve’ their use for translation of sub-genomic RNA. It was also shown in vitro that N transfection into cells resulted in multi-nucleate cells, indicating the delay of cytokinesis (24). This would provide favorable and prolonged conditions for the virus intracellularly to continue to synthesize its genome and sub-genome, translate its proteins and enable sufficient virion packaging. Moreover, N is proposed to dampen the host cell’s antiviral transcriptional response within the nucleus (8). Nevertheless, confirming the presence SARS-CoV-2 N in the nucleolus and understanding its role would elucidate the pathogenicity of this virus.
N is an essential component of newly formed virions as it ensures a proper ‘delivery’ of the replicated viral RNA genome within the developing envelope (28, 29). Moreover, it is essential for proper viral RNA dependent RNA polymerase activity, as demonstrated in Influenza A (29). As such, targeting the activity of N would offer a potent antiviral activity against SARS-CoV-2. In fact, N was shown to be an effective anti-viral target against Influenza A. One of the useful properties of N is its numerous binding sites, which have been shown to accommodate various drugs (29, 30). For example, compounds which can target the tail-loop binding pocket abrogate N oligomerization, while the compound F66 binds to the RNA-binding groove of the protein and is associated with improved survival in animal models infected with Influenza A (29). Figure 1 illustrates how the N of SARS-CoV-2 facilitates virus replication and mitigates the host cell response, thus further strengthening its position as a promising target of anti-viral drugs.

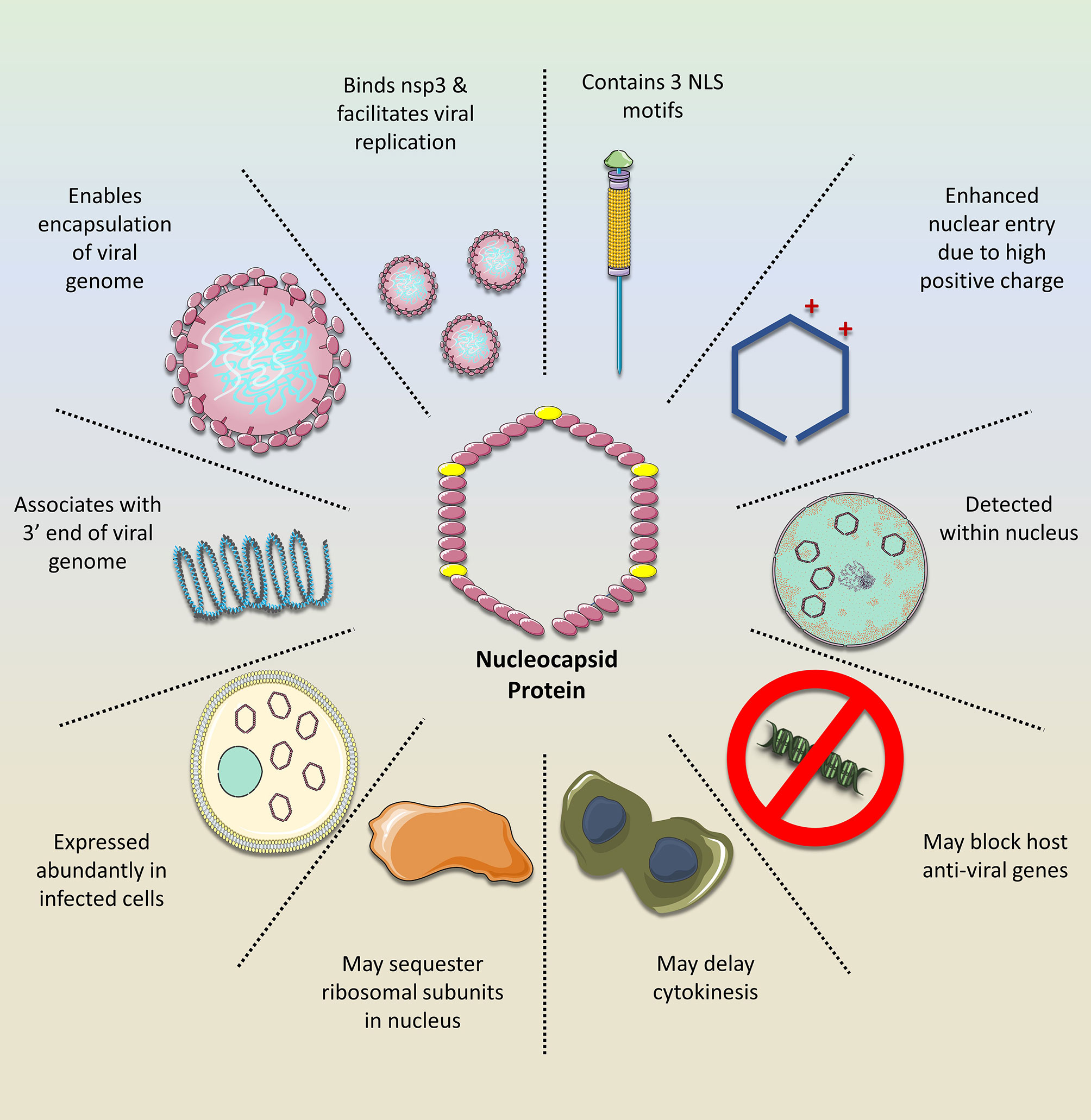
Figure 1 The importance of the SARS-CoV-2 nucleocapsid protein (N). The N exerts numerous functions that facilitate viral replication while mitigating the host cell response. Owing to its NLS motifs, the protein retains a relatively high positive charge, compared to the N of other coronavirus clades. This enhances its transport into the nucleus where it may silence host anti-viral genes while sequestering ribosomal subunits, possibly for viral mRNA translation, as demonstrated with the N of other related viruses. Moreover, the N is important for stabilizing the interaction between the viral mRNA and nsp3 protein, which facilitates genome replication. In addition, it tethers the newly emerged viral RNA to the viral envelope, ultimately allowing for its encapsulation and formation of new viral progeny. Given these features and its abundance in the infected cell, it would be a promising drug target against SARS-CoV-2.
Ivermectin
The Discovery of Ivermectin
IVM was originally discovered from organisms that were isolated from soil samples collected from the woods nearby to Kitasato Institute in Kawana, Japan. Fermentation products released by a bacterium from the soil, which was later classified as Streptomyces acermitilis, appeared to exhibit antiparasitic activity (specifically against Nematospiroides dubius). Purification and isolation of the bioactive compounds showed naturally occurring macrocyclic lactones, and these were subsequently named avermectins. Avermectins are made up of four compounds, which exist as two variants: A1, A2, B1, and B2. Variants ‘A’ and ‘B’ indicate the presence of methoxy or hydroxyl groups, respectively, at the C5 position. Number ‘1’ describes the double bond between C22 and C23. On the other hand, number ‘2’ indicates the presence of hydrogen at C22 and a hydroxyl group at C23. B1 avermectins were proven to be most active on oral administration, and on this basis, IVM was chemically derived. IVM contains an 80:20 combination of 22,23-dihydro-acvermectin B1a and 22,23-dihydro-avermectin B1b. Its antiparasitic effects are primarily caused by high-affinity irreversible binding to glutamate-gated chloride (Cl-) channels located on nerve and muscle cells of nematode, which leads to hyperpolarization (9, 31). Ultimately, the increased permeability to Cl- results in paralysis and death of the nematode (31).
As of yet, IVM has treated hundreds of millions of people with onchocerciasis, most commonly given at 150-200 μg/kg of body weight for one dose initially, and repeated at 6-12 monthly intervals as appropriate (32). Its use extends to a broad spectrum of parasitic nematodes on both oral and parenteral administration, and is also effective against arthropods, including lice (33).
Importantly, IVM was approved by the FDA for human use in 1987 (34). Its low toxicity and safety are attributed to the fact that its human target receptors are ‘secluded’ in the CNS, and IVM has not been shown to cross the blood-brain barrier. In addition, IVM displays a 100-fold greater affinity for parasitic Cl- channels compared to the human homologs (35). Moreover, severe detrimental effects in humans were shown only in those who over-dosed using approximately 15.4 mg/kg body weight IVM, which is 77 times above the prescribed dose. This corroborates the advantage of repurposing drugs, as these medications have already been tested arduously and extensively to confirm their efficacy and safety, thereby decreasing the transit time from shelf to intake.
Screening for Inhibitors of Nuclear Import
The potential of IVM as an inhibitor of nuclear transport of viral proteins was initially suggested in 2011. Initially, Wagstaff and colleagues screened for nuclear import inhibitors, which block the interaction between IMPs and potential target cellular proteins (21). They randomly selected 480 compounds from LOPAC1280 (Library of Pharmacologically Active Compounds; Sigma, St. Louis, MO). IVM surfaced as a drug that generally inhibits IMP activity (21). A year later, they confirmed that this apparent activity of IVM also inhibits nuclear transport of viral proteins HIV and Dengue virus in HeLa cells (36). Specifically, it was shown that GFP-tagged IMP was significantly reduced in the nucleus of HeLa cells after 3 hrs. of co-incubation with IVM (36). Moreover, the effect was unique to IMPα/β interactions and did not affect proteins bound only to IMPβ1. The importance of blocking the nuclear import of viral proteins emerged when it was later shown that IVM also prevented replication of HIV (36). As such, it surfaced as a possible repurposed drug, capable of preventing viral cargo from interacting with IMPα/β for nuclear import, with the potential to result in viral ‘death’ (21, 36).
Soon after, the effect of IVM against the nuclear import of viral proteins was further validated. For example, IVM prevented nuclear translocation of nsp5 in Dengue virus, West Nile virus, and influenza and inhibited transport of large tumor antigen (T-ag) in simian virus (25, 37, 38). The Wagstaff et al., 2011 and 2012 studies were pivotal in providing much of the initial rationale for the recent consideration of IVM as a SARS-CoV-2 antiviral agent. In fact, it was the same researchers who nine years later, in 2020, demonstrated that IVM inhibits SARS-CoV-2 in vitro replication (8).
Given its efficacy in inhibiting nuclear import of other viral proteins, the anti-viral effect of IVM against SARS-CoV-2 was evaluated shortly after the pandemic erupted (8). Specifically, Vero/hSLAM cells were inoculated with SARS-CoV-2 isolate for 2 hrs., followed up with supplementation of 5 μM of IVM. Within 24 hrs. after treatment, there was a 93% reduction of viral RNA in the supernatant and 99.8% reduction of cellular viral RNA, compared to controls. After 48 hrs., there was a further 5000-fold reduction of viral RNA in the supernatant as well as the cell pellets, indicating that cells were essentially ‘cleared’ of SARS-CoV-2 (8). Although IVM possessed a potent antiviral activity (IC50= ~2μM), no cytotoxicity was detected at any time points in this study (8).
IVM Specifically Interacts With IMPα
IVM was shown to specifically inhibit IMP α/β mediated nuclear import required for replication of HIV-1 and Dengue virus, and therefore it was proposed as the potential mechanism by which it inhibits SARS-CoV-2 (36). Indeed, this baton was passed on, and a subsequent study verified the IVM-IMPα interaction in host cells (38). In fact, it was shown that IVM not only inhibits IMPα association with IMPβ, but can even dissociate IMPα/β heterodimers (38). The specific binding target of IVM was identified, using CD spectroscopy, to be the alpha-helical rich ‘armadillo’ (ARM) domain of IMPα. Moreover, as concentrations of IVM increased, alpha helices in the ARM domain became increasingly destabilized. No changes were detected in the structure of IMPβ. They further verified that this observed effect on IMPα impaired its binding to NLS-containing nsp5 from Dengue Virus (38). As such, preventing N interaction with IMPα, is a likely mechanism that contributes to IVM’s ability to hinder SARS-CoV-2 in vitro replication (Figure 2).

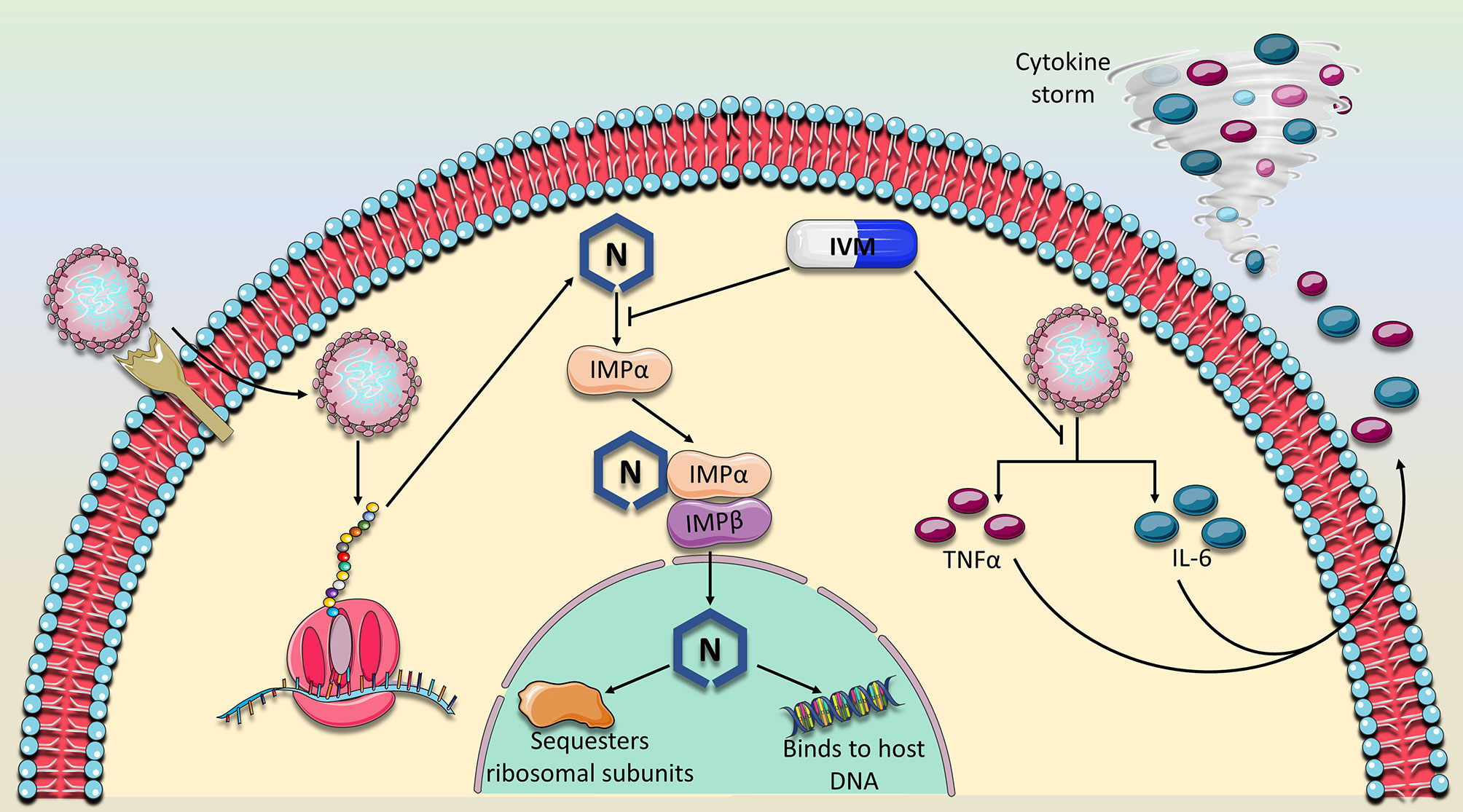
Figure 2 Proposed mechanism of action of Ivermectin against SARS-CoV-2. IVM has previously been established as a nuclear import inhibitor by binding to and antagonizing the ability of the importin (IMPα) to bind to its target cargo. Because the nucleocapsid (N) protein contains a nuclear localization signal, IVM is expected to prevent the binding of IMPα to the N binding site. Consequently, N would not perform its nuclear activity which is thought to suppress the host immune response and sequester ribosomal subunits, mechanisms which are thought to abrogate sufficient viral replication. In addition, the expression of two major cytokines, TNFα and IL-6 which drive the detrimental cytokine storm in COVID-19 patients were also shown to be dampened in the presence of IVM. As of yet, these two major mechanisms which involve viral replication and immune response suppression appear to characterize the main activities of IVM against SARS-CoV-2.
The Implications of Disrupting IMPα Activity for the Host Cells
Because IVM emerged as a general inhibitor of IMPα-dependent nuclear cargo, it is important to consider the implications this may have on host cell proteins and functions. However, any effect would likely be non-detrimental given the safety record of IVM over the past 50 years and its transient prescription for an acute disease (31).
Notably, expression levels of IMPα vary in a cell and developmental specific manner, particularly during differentiation processes (39). Animal knock-out studies for impα, highlighted its essential role in reproductive organ development. Specifically, impa-/- mice developed lower reproductive organ function in females, including insufficient follicles’ growth during the maturation stage in the ovaries, incomplete uterus construction, and reduced serum progesterone (40). Moreover, estrogen-responsive genes were also not efficiently expressed, indicating IMPα may be involved in hormonal regulation. Other cells like muscle stem cells underwent apoptosis and depletion (39).
IVM was also shown to disrupt the oxygen regulatory mechanisms (41). Hypoxia-induced transcription factors (HIFs) regulate cellular adaptation to decreased oxygenation within the cell. Hypoxia renders the HIF subunit, HIFα stable and causes it to accumulate within the nucleus where it induces transcription of genes that may readjust oxygen levels. HIFα translocation into the nucleus requires nuclear import in an NLS-IMPα/β dependent manner. Indeed, it was shown that IVM results in decreased association between HIFα and IMPα, preventing its path into the nucleus. Subsequently, nuclear HIFα and transcription of target oxygen-regulatory genes was reduced (41).
Pharmacokinetic studies conducted by MERCK show that IVM plasma concentrations peak after 4 hours, following 12 mg doses in healthy human volunteers (42). Subsequently, it is metabolized in the liver and its break down products are mainly excreted in the feces over a period of 12 days. Its half-life is around 18 hrs. Moreover, it was shown that it does not bind permanently to its target proteins.
Other Possible Modes of Antiviral Activity by IVM
A recent molecular docking study demonstrated that in addition to IMPα, IVM showed high binding affinity to the viral RNA-dependent RNA polymerase (RdRp) complexed with RNA helicase compared to other 10 viral targets included in the analysis (43). However, it was later shown that IVM does not bind to viral RdRp in both Zika virus (Z.V.) and Dengue virus (38). It remains to be identified if IVM may bind to RdRp in coronaviruses.
Other mechanisms of IVM action have also been identified (Figure 2). For example, it previously was shown to suppress the production of Interleukin-6 (IL-6) and Tumor Necrosis Factor alpha (TNFα), two major components of the detrimental cytokine storm induced by SARS-CoV-2 (44). Moreover, a study in Syrian hamsters showed that IVM did not affect SARS-CoV-2 viral load but overall dramatically reduced IL-6/IL-10 ratio and modulated infection outcomes (45). Specifically, hamsters that were inoculated with SARS-CoV-2 were subcutaneously injected with IVM (0.4 mg/kg body weight). IVM reduced severity of clinical symptoms in males, but completely eliminated symptoms in females, which suggests a gender-specific effect of this drug and a factor that should be considered in clinical trials. The gender-specific modulation of IVM on cytokines was also apparent. While females displayed lower levels of cytokines such as IL-6, INFγ and TNFα, males on the other hand developed an enhanced production of INFγ. Notably, viral load in nasal and lung tissues, as well as viral replication rate were not altered in either gender after administration of IVM (45). This is in contrast to the finding that IVM significantly blocks viral replication in vitro and it may be attributed to the much higher dose of IVM that was used (8). However, it is important to note that the dose of IVM that was used on the cells (5 μM) is approximately 50-fold higher than the normal Cmax associated with one dose of IVM (200 μg/kg) (46, 47). Therefore, it is important to establish a dose-dependent effect of IVM on viral load and safety in human COVID-19 patients at various doses.
Further, IVM was shown to induce an elevated level of IL-6 and TNFα in onchocerciasis patients, two days after a single dose (150 μg/kg body weight) (48). However, this was attributed to the destruction of the parasite microfilariae, which would usually not be a factor in COVID-19 patients.
Thus far, studies on IVM highlight that it remains important to identify the specific dose of IVM that may reduce viral load, without adverse effects, in humans and to understand if it will differentially affect male and female COVID-19 patients.
Adverse Effects Reported in Animals and Humans in Previous Studies Using Ivermectin
The direct toxic effects of IVM were first identified in animal studies, mainly as an antiparasitic treatment. The vast amount of evidence around the use of IVM exists using dose regimens of 150-200 μg/kg of body weight. Hence, the risks and associated side effects are mostly reported at these doses. Studies suggest the common adverse effects are rash, headache, nausea and dizziness, while transient tachycardia is rare and self-limiting (49). Other effects include ataxia, sweating, tremors, and in some cases, coma and death (50).
A retrospective study looking at residents of an extended care facility showed increased rates of death in patients treated with IVM for resistant scabies. These study results were criticized due to some significant limitations of the study, and therefore, the deaths of these residents could not be reliably attributed to the IVM. For example, there was no control of the lasting previous drugs used to treat the scabies, some of which are known for their toxic effects. Importantly, IVM’s toxic effects are short term and are usually resolved (51).
Studies exploring the adverse event profiles of patients on high doses of IVM have also been conducted. Higher dose levels (300-1000 μg/kg) were administered to healthy individuals with head lice, as part of a double-blind and randomized trial, with adverse events reported as having no clinical or biochemical significance (50).
Clinical Trials
The Effect of Ivermectin on SARS-CoV-2 Patients
Soon after IVM emerged as a potential therapeutic agent, clinical trials on COVID-19 patients ensued. However, the available published data and ongoing clinical trials, which are summarized in Table 1, do not provide a clear and uniform understanding of the effect of IVM on COVID-19 patients. This is mainly due to small sample sizes (n=12-203) and the lack of information specifying when exactly IVM is administered after testing positive for SARS-CoV-2 (46, 52–54). It is important to highlight how soon after testing positive the patient receives IVM, in addition to the degree of COVID-19 severity, in order to understand if the effect of the drug is dependent on time and symptom severity. Additionally, several studies are retrospective in which investigators examined past COVID-19 patients who were prescribed IVM, without proper placebo control groups (46, 53). Moreover, most of the studies utilize the antiparasitic effective dose for IVM (0.2 mg/kg body weight), which is substantially less than the equivalent in vitro dose of IVM used against SARS-CoV-2 (8, 53, 54). Nevertheless, the available data does indicate that IVM may, in fact, be effective against COVID-19.
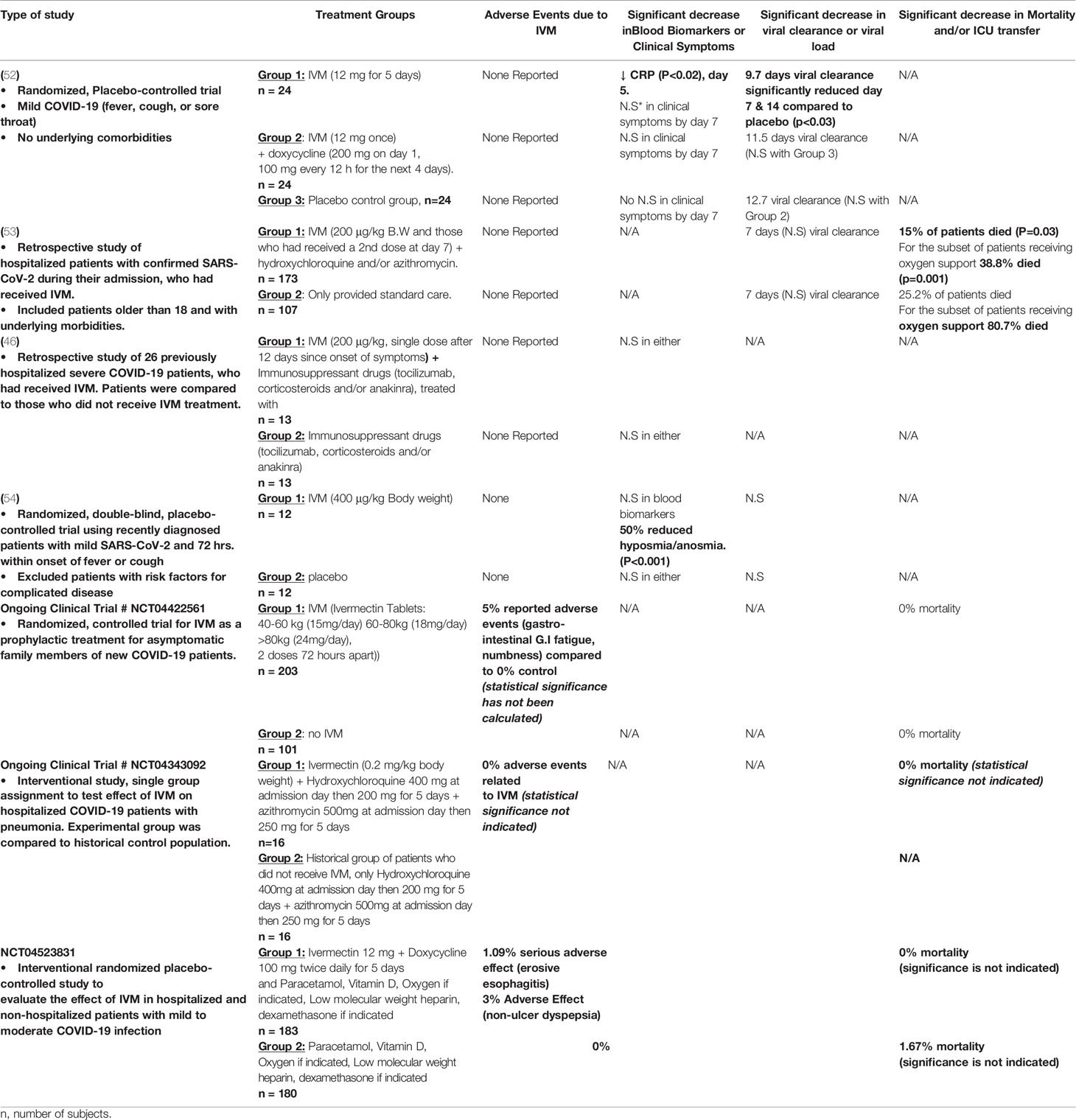
Table 1 Outcomes in Current Clinical Trials at Ivermectin.
One of the first published studies involved a randomized, controlled double-blind study on 72 hospitalized COVID-19 with mild symptoms (52). Patients that were admitted to the hospital within the last 7 days were either treated with IVM alone (12 mg for 5 days), IVM and doxycycline (12 mg for 1 day, 200 mg doxycycline on day 1 and 100 mg doxycycline every 12 hrs. for days 2-6), or with placebo. The most significant effect of IVM was detected for the rate of viral clearance, measured by a negative rRT-PCR on nasopharyngeal swab. Specifically, the 5-day IVM treatment group demonstrated the fastest rate of viral clearance (approximately 10 days; p<0.02), compared to placebo (approximately 13 days). However, there was no significant difference between the groups for symptoms like cough, sore throat and fever and adverse drug effects (52). Limitations for this study include the exclusion of patients with underlying morbidities and lack of follow-up for mortality and ICU transfers after day 7.
Another recently published clinical study in Florida involved 280 hospital-admitted patients who developed COVID-19 during admission (53). However, it was based on a retrospective analysis of patients and therefore lacked adequate controls. Patients were grouped according to whether or not they received a single dose of IVM (200 μg/kg body weight) and standard care (hydroxychloroquine and azithromycin, unspecified dose) or those who only received standard care. It was not stated, though, at which day post-PCR testing that patients received the drug. The most prominent effect of IVM was the reduction in mortality in the treatment group (15% mortality, p= 0.03) compared to the control group (25% mortality). Mortality was especially reduced in the severe subgroup of patients receiving oxygen support (38.% mortality in IVM group, compared to 80.7% mortality in non-IVM group; p = 0.001). In fact, hospital stay was similar in both groups, as was the rate of viral clearance. However, data was lacking for clinical symptoms as it was not a main outcome (53). In contrast to the previous study, this trial included patients with comorbidities and no adverse drug events were reported. Although this study is limited because it is retrospective, it suggests that IVM may be beneficial in reducing mortality almost by one half, especially for patients receiving oxygen support. Nevertheless, it remains ambiguous as to how soon after testing positive for SARS-CoV-2, patients received IVM, which would have been an important guideline. The results of the few addition published IVM trials are summarized in Table 1.
There are currently around 50 clinical trials taking place registered on clinicaltrials.gov, which study the effect of IVM as a prophylactic or therapeutic drug (35). Five studies have completed testing at Phase 1, 2 or 3 and three have posted their real-time results, which are included in Table 1. One of the most promising outcomes include 0% mortality in COVID-19 patients who developed pneumonia (National Clinic Trial Number NCT04343092), however there is no report of an adequate control group and its corresponding rate of mortality. Another ongoing trial (NCT04523831) on COVID-19 hospitalized and non-hospitalized patients also demonstrated 0% mortality, in the IVM group, compared to 1.67% mortality in the control group. Like published studies, ongoing clinical trials also do not present thorough outcomes and the significance statistics are still lacking. As such, more trials are needed which include proper placebo control groups, testing of various doses and records of numerous outcomes.
Ivermectin Compared to Other Anti-SARS-CoV-2 Drugs
In addition to IVM, many clinical trials have been conducted to test drugs for COVID-19, many of which have been concluded (47). Most of these medications did not result in significantly improved outcomes, however a few drugs were associated with slightly beneficial effects.
Remdesivir (RMV) has previously been shown to target the viral RNA dependent RNA polymerase in SARS-CoV-1 (55). In the context of SARS-CoV-2, RMV prescribed once daily for 10 days (200 mg day 1, 100 mg days 9-10), showed a survival benefit by days 15 and 28 in patients who did not require oxygen support (56). On the other hand, a large global study by the WHO did not find any clinical benefit for RMV (57). However, an in vitro study suggests a possible synergistic effect of combined RMV and IVM. Specifically, EC50 of approximately 2.3 μM IVM and 1.9 μM RMV were shown to disrupt viral cytopathic activity (58). Because RMV is FDA-approved for the treatment of COVID-19, it is warranted to explore its effect in combination with IVM in clinical trials.
Dexamethasone (6 mg for 10 days) decreased mortality only in severe cases requiring oxygen and mechanical ventilation but was ineffective for mild cases and did not result in any adverse effects (59). Interestingly, a clinical trial (NCT04425863) involving a combined therapy of IVM (0.6 mg/mL solution), dexamethasone (4 mg injection), aspirin (250 mg tablets) and enoxaparin (injection) did indicate a favorable outcome. All patients with mild COVID-19 symptoms (n=135) fully recovered and their symptoms did not worsen. Of those who entered the study displaying severe symptoms (n= 31), one patient perished.
The use of convalescent plasma is also not entirely promising as preliminary analysis based on 1873 reported deaths among 10,406 randomized patients, shows no significant difference in the primary endpoint of 28-day mortality (18% convalescent plasma vs. 18% usual care alone, p=0.34). Although some early studies showed some clinical benefits for convalescent plasma in COVID-19 patients (47), a recent press release from the largest randomized clinical trial, known as the ‘RECOVERY Trial’, revealed otherwise (60). The investigators concluded no evidence of benefit for convalescent plasma in treatment of COVID-19, whereby the 28-day mortality did not differ significantly between the treatment and the control groups. Recently two clinical trials showed that monoclonal antibodies against the spike protein can disrupt progression of early COVID-19 infection (61, 62). However, this type of therapeutic remains very expensive and largely unavailable.
Finally, a recent study, still awaiting peer-review, demonstrated that treatment with the IL-6 receptor antagonists, tocilizumab and sarilumab, improved the clinical outcome including survival in critically ill COVID-19 patients (63). However, these drugs remain expensive and not widely available especially in poor and developing countries.
Concluding Remarks and Perspectives
The available data from IVM clinical trials lack uniformity and have not established the optimal anti-viral dose. However, the evidence does support its safety and efficacy in improving survival rates, especially compared to the other aforementioned drugs. It is important to note that past research has demonstrated the importance of combined, rather than anti-viral monotherapy. Indeed, the use of a single drug does not efficiently suppress long-term replication of the virus (64). As evident by the ongoing clinical trials for the treatment of COVID-19, the most efficient decrease in mortality (0%) was largely a result of multiple prescribed drugs including IVM, hydroxychloroquine and azithromycin or IVM and doxycyline Table 1. Given the wide use of numerous drugs to treat COVID-19 patients, it remains imperative to explore the optimal combination of various therapies.
Notably, the clinical outcomes upon prescribing IVM on its own did not result in significantly improved outcomes for COVID-19 patients and nor should it be particularly encouraged (54). In fact, cross-resistance to other medications may be induced as a result of selective pressure resulting from a single medication (64). This may be a likely event as RNA viruses are well noted for their pronounced capacity for mutations, a finding which has already been established also for SARS-CoV-2 (65). Therefore, although IVM may contribute to the suppression of SARS-CoV-2 replication, it is important not to dismiss the risk of selecting for highly pathological and resistant viral strains when using a sole medication. That said, in a recent clinical trial that we have just concluded and is under review, we show that a single dose of IVM can significantly reduce the viral load in asymptomatic SARS-CoV-2 positive subjects. However, in these subjects, zinc and vitamin C were concomitantly used.
The available data thus far suggests a favorable outcome when using IVM in specific doses and in particular drug combinations. It remains imperative to establish the most effective doses, combination, and timing of drug administration as it may largely determine the therapeutic outcome. Although vaccines are currently being distributed, they do not guarantee complete protection against SARS-CoV-2. Therefore, it is important to establish therapeutic alternatives in the event that viral re-infection occurs. Given the promising emerging clinical data from IVM studies and the unprecedented public health threat that the pandemic poses, it is critical that further specific and well-designed studies are carried out to validate the therapeutic potential of IVM.
Author Contributions
AE generated the concept. ZW and MW wrote the first draft. All authors revised the manuscript and approved it before submission. HY generated funding.
Funding
This study was supported by Qatar University Grants # QUCG-BRC-20_21 and QUHI-BRC-20/21-1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wang Y, Grunewald M, Perlman S. Coronaviruses: An Updated Overview of Their Replication and Pathogenesis. Methods Mol Biol (2020) 2203:1–29. doi: 10.1007/978-1-0716-0900-2_1
2. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 2:271–80. doi: 10.1016/j.cell.2020.02.052
3. Lu G, Wang Q, Gao GF. Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol (2015) 23(8):468–78. doi: 10.1016/j.tim.2015.06.003
4. Mathew S, Faheem M, Hassain NA, Benslimane FM, Thani AAA, Zaraket H, et al. Platforms Exploited for SARS-CoV-2 Vaccine Development. Vaccines (Basel) (2020) 9(1):11. doi: 10.3390/vaccines9010011
5. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
6. Chen J, Wang R, Wang M, Wei GW. Mutations Strengthened SARS-CoV-2 Infectivity. J Mol Biol (2020) 432(19):5212–26. doi: 10.1016/j.jmb.2020.07.009
7. Singh PK, Kulsum U, Rufai SB, Mudliar SR, Singh S. Mutations in SARS-CoV-2 Leading to Antigenic Variations in Spike Protein: A Challenge in Vaccine Development. J Lab Physicians (2020) 12(2):154–60. doi: 10.1055/s-0040-1715790
8. Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res (2020) 178:104787. doi: 10.1016/j.antiviral.2020.104787
9. Crump A, Ōmura S. Ivermectin, ‘wonder drug’ from Japan: the human use perspective. Proc Jpn Acad Ser B Phys Biol Sci (2011) 87(2):13–28. doi: 10.2183/pjab.87.13
10. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells (2020) 9(5):1267. doi: 10.3390/cells9051267
11. Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol (2000) 81(Pt 4):853–79. doi: 10.1099/0022-1317-81-4-853
12. Cong Y, Ulasli M, Schepers H, Mauthe M, V’kovski P, Kriegenburg F, et al. Nucleocapsid Protein Recruitment to Replication-Transcription Complexes Plays a Crucial Role in Coronaviral Life Cycle. J Virol (2020) 94(4):e01925–19. doi: 10.1128/JVI.01925-19
13. Sawicki SG, Sawicki DL. A new model for coronavirus transcription. Adv Exp Med Biol (1998) 440:215–9. doi: 10.1007/978-1-4615-5331-1_26
14. McBride R, van Zyl M, Fielding BC. The coronavirus nucleocapsid is a multifunctional protein. Viruses (2014) 6(8):2991–3018. doi: 10.3390/v6082991
15. Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun (2020) 11(1):6041. doi: 10.1038/s41467-020-19843-1
16. Fulcher AJ, Jans DA. Regulation of nucleocytoplasmic trafficking of viral proteins: an integral role in pathogenesis? Biochim Biophys Acta (2011) 1813(12):2176–90. doi: 10.1016/j.bbamcr.2011.03.019
17. Rout MP, Aitchison JD. Pore relations: nuclear pore complexes and nucleocytoplasmic exchange. Essays Biochem (2000) 36:75–88. doi: 10.1042/bse0360075
18. Fried H, Kutay U. Nucleocytoplasmic transport: taking an inventory. Cell Mol Life Sci (2003) 60(8):1659–88. doi: 10.1007/s00018-003-3070-3
19. Goldfarb DS, Corbett AH, Mason DA, Harreman MT, Adam SA. Importin alpha: a multipurpose nuclear-transport receptor. Trends Cell Biol (2004) 14(9):505–14. doi: 10.1016/j.tcb.2004.07.016
20. Weis K. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell (2003) 112(4):441–51. doi: 10.1016/s0092-8674(03)00082-5
21. Wagstaff KM, Rawlinson SM, Hearps AC, Jans DA. An AlphaScreen®-based assay for high-throughput screening for specific inhibitors of nuclear import. J Biomol Screen (2011) 16(2):192–200. doi: 10.1177/1087057110390360
22. Gussow AB, Auslander N, Faure G, Wolf YI, Zhang F, Koonin EV. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc Natl Acad Sci USA (2020) 117(26):15193–9. doi: 10.1073/pnas.2008176117
23. Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep (2000) 1(5):411–5. doi: 10.1093/embo-reports/kvd092
24. Wurm T, Chen H, Hodgson T, Britton P, Brooks G, Hiscox JA. Localization to the nucleolus is a common feature of coronavirus nucleoproteins, and the protein may disrupt host cell division. J Virol (2001) 75(19):9345–56. doi: 10.1128/JVI.75.19.9345-9356.2001
25. Gotz V, Magar L, Dornfeld D, Giese S, Pohlmann A, Hoper D, et al. Influenza A viruses escape from MxA restriction at the expense of efficient nuclear vRNP import. Sci Rep (2016) 6:23138. doi: 10.1038/srep23138
26. Zeng W, Liu G, Ma H, Zhao D, Yang Y, Liu M, et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem Biophys Res Commun (2020) 527(3):618–23. doi: 10.1016/j.bbrc.2020.04.136
27. You JH, Reed ML, Hiscox JA. Trafficking motifs in the SARS-coronavirus nucleocapsid protein. Biochem Biophys Res Commun (2007) 358(4):1015–20. doi: 10.1016/j.bbrc.2007.05.036
28. Hurst KR, Koetzner CA, Masters PS. Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. J Virol (2013) 87(16):9159–72. doi: 10.1128/JVI.01275-13
29. Hu Y, Sneyd H, Dekant R, Wang J. Influenza A Virus Nucleoprotein: A Highly Conserved Multi-Functional Viral Protein as a Hot Antiviral Drug Target. Curr Top Med Chem (2017) 17(20):2271–85. doi: 10.2174/1568026617666170224122508
30. Lin SM, Lin SC, Hsu JN, Chang CK, Chien CM, Wang YS, et al. Structure-Based Stabilization of Non-native Protein-Protein Interactions of Coronavirus Nucleocapsid Proteins in Antiviral Drug Design. J Med Chem (2020) 63(6):3131–41. doi: 10.1021/acs.jmedchem.9b01913
31. Ikeda T. Pharmacological effects of ivermectin, an antiparasitic agent for intestinal strongyloidiasis: its mode of action and clinical efficacy. Nihon Yakurigaku Zasshi (2003) 122(6):527–38. doi: 10.1254/fpj.122.527
32. González Canga A, Sahagún Prieto AM, Diez Liébana MJ, Fernández Martínez N, Sierra Vega M, García Vieitez JJ. The pharmacokinetics and interactions of ivermectin in humans–a mini-review. AAPS J (2008) 10(1):42–6. doi: 10.1208/s12248-007-9000-9
33. Schröder J, Swan GE, Soll MD, Hotson IK. Efficacy of ivermectin against ectoparasites of cattle in South Africa. J S Afr Vet Assoc (1985) 56(1):31–5.
34. Juarez M, Schcolnik-Cabrera A, Dueñas-Gonzalez A. The multitargeted drug ivermectin: from an antiparasitic agent to a repositioned cancer drug. Am J Cancer Res (2018) 8(2):317–31.
35. Kaur H, Shekhar N, Sharma S, Sarma P, Prakash A, Medhi B. Ivermectin as a potential drug for treatment of COVID-19: an in-sync review with clinical and computational attributes. Pharmacol Rep (2021). doi: 10.1007/s43440-020-00195-y
36. Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J (2012) 443(3):851–6. doi: 10.1042/BJ20120150
37. Tay MY, Fraser JE, Chan WK, Moreland NJ, Rathore AP, Wang C, et al. Nuclear localization of dengue virus (DENV) 1-4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antiviral Res (2013) 99(3):301–6. doi: 10.1016/j.antiviral.2013.06.002
38. Yang SNY, Atkinson SC, Wang C, Lee A, Bogoyevitch MA, Borg NA, et al. The broad spectrum antiviral ivermectin targets the host nuclear transport importin α/β1 heterodimer. Antiviral Res (2020) 177:104760. doi: 10.1016/j.antiviral.2020.104760
39. Oka M, Yoneda Y. Importin α: functions as a nuclear transport factor and beyond. Proc Jpn Acad Ser B Phys Biol Sci (2018) 94(7):259–74. doi: 10.2183/pjab.94.018
40. Moriyama T, Nagai M, Oka M, Ikawa M, Okabe M, Yoneda Y. Targeted disruption of one of the importin α family members leads to female functional incompetence in delivery. FEBS J (2011) 278(9):1561–72. doi: 10.1111/j.1742-4658.2011.08079.x
41. Kosyna FK, Nagel M, Kluxen L, Kraushaar K, Depping R. The importin α/β-specific inhibitor Ivermectin affects HIF-dependent hypoxia response pathways. Biol Chem (2015) 396(12):1357–67. doi: 10.1515/hsz-2015-0171
42. TABLETS; STROMECTOL®(IVERMECTIN) (MERCK). Available at: https://www.merck.com/product/usa/pi_circulars/s/stromectol/stromectol_pi.pdf.
43. Sen Gupta PS, Biswal S, Panda SK, Ray AK, Rana MK. Binding mechanism and structural insights into the identified protein target of COVID-19 and importin-α with. J Biomol Struct Dyn (2020) 1–10. doi: 10.1080/07391102.2020.1839564
44. Zhang X, Song Y, Ci X, An N, Ju Y, Li H, et al. Ivermectin inhibits LPS-induced production of inflammatory cytokines and improves LPS-induced survival in mice. Inflamm Res (2008) 57(11):524–9. doi: 10.1007/s00011-008-8007-8
45. de Melo GD, Lazarini F, Larrous F, Feige L, Kergoat L, Marchio A, et al. Anti-COVD-19 efficacy of ivermectin in the golden hamster. BioRxiv (2020). doi: 10.1101/2020.11.21.392639
46. Camprubí D, Almuedo-Riera A, Martí-Soler H, Soriano A, Hurtado JC, Subirà C, et al. Lack of efficacy of standard doses of ivermectin in severe COVID-19 patients. PloS One (2020) 15(11):e0242184. doi: 10.1371/journal.pone.0242184
47. Kaddoura M, AlIbrahim M, Hijazi G, Soudani N, Audi A, Alkalamouni H, et al. COVID-19 Therapeutic Options Under Investigation. Front Pharmacol (2020) 11:1196. doi: 10.3389/fphar.2020.01196
48. Njoo FL, Hack CE, Oosting J, Luyendijk L, Stilma JS, Kijlstra A. C-reactive protein and interleukin-6 are elevated in onchocerciasis patients after ivermectin treatment. J Infect Dis (1994) 170(3):663–8. doi: 10.1093/infdis/170.3.663
49. Guzzo CA, Furtek CI, Porras AG, Chen C, Tipping R, Clineschmidt CM, et al. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J Clin Pharmacol (2002) 42(10):1122–33. doi: 10.1177/009127002401382731
51. Alexander ND, Bockarie MJ, Kastens WA, Kazura JW, Alpers MP. Absence of ivermectin-associated excess deaths. Trans R Soc Trop Med Hyg (1998) 92(3):342. doi: 10.1016/s0035-9203(98)91035-5
52. Ahmed S, Karim MM, Ross AG, Hossain MS, Clemens JD, Sumiya MK, et al. A five-day course of ivermectin for the treatment of COVID-19 may reduce the duration of illness. Int J Infect Dis (2020) 103:214–6. doi: 10.1016/j.ijid.2020.11.191
53. Rajter JC, Sherman MS, Fatteh N, Vogel F, Sacks J, Rajter JJ. Use of Ivermectin Is Associated With Lower Mortality in Hospitalized Patients With Coronavirus Disease 2019: The Ivermectin in COVID Nineteen Study. Chest (2021) 159(1):85–92. doi: 10.1016/j.chest.2020.10.009
54. Chaccour C, Ruiz-Castillo P, Richardson MA, Moncunill G, Casellas A, Carmona-Torre F, et al. The SARS-CoV-2 Ivermectin Navarra-ISGlobal Trial (SAINT) to Evaluate the Potential of Ivermectin to Reduce COVID-19 Transmission in low risk, non-severe COVID-19 patients in the first 48 hours after symptoms onset: A structured summary of a study protocol for a randomized control pilot trial. Trials (2020) 21(1):498. doi: 10.1186/s13063-020-04421-z
55. Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio (2018) 9(2):e00221-18. doi: 10.1128/mBio.00221-18
56. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med (2020) 383(19):1813–26. doi: 10.1056/NEJMoa2007764
57. Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, Abdool Karim Q, et al. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N Engl J Med (2020) 384:497–511. doi: 10.1056/NEJMoa2023184
58. Jeffreys L, Pennington SH, Duggan J, Breen A, Jinks J, Ardrey A, et al. Remdesivir-Ivermectin combination displays synergistic interaction with improved in vitro antiviral activity against SARS-CoV-2. (2020). doi: 10.1101/2020.12.23.424232
59. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N Engl J Med (2020) 384:693–704. doi: 10.1056/NEJMoa2021436
60. Recovery Trial Closes Recruitment to Convalescent Plasma Treatment for Patients Hospitalised with COVID-19. Nuffield Department of Population Health. University of Oxford (2021).
61. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a Neutralizing Antibody CocktailOutpatients with Covid-19. N Engl J Med (2021) 384(3):238–51. doi: 10.1056/NEJMoa2035002
62. Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med (2021) 384(3):229–37. doi: 10.1056/NEJMoa2029849
63. Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, Arabi YM, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19 –Preliminary report. N Engl J Med (2021). doi: 10.1056/NEJMoa2100433
64. Larder BA. Viral resistance and the selection of antiretroviral combinations. J Acquir Immune Defic Syndr Hum Retrovirol (1995) 10(Suppl 1):S28–33. doi: 10.1097/00042560-199510001-00007
Keywords: COVID-19, SARS-CoV-2, ivermectin, coronavirus, mechanism of action
Citation: Wehbe Z, Wehbe M, Iratni R, Pintus G, Zaraket H, Yassine HM and Eid AH (2021) Repurposing Ivermectin for COVID-19: Molecular Aspects and Therapeutic Possibilities. Front. Immunol. 12:663586. doi: 10.3389/fimmu.2021.663586
Received: 03 February 2021; Accepted: 15 March 2021;
Published: 30 March 2021.
Edited by:
Shuofeng Yuan, The University of Hong Kong, Hong KongReviewed by:
Jun Wang, University of Arizona, United StatesShailesh Kumar Patel, Chhattisgarh Kamdhenu Vishwavidyalaya, India
Copyright © 2021 Wehbe, Wehbe, Iratni, Pintus, Zaraket, Yassine and Eid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hadi M. Yassine, hyassine@qu.edu.qa; Ali H. Eid, ali.eid@qu.edu.qa
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7251046/
Ivermectin, antiviral properties and COVID-19: a possible new mechanism of action

Abstract
Ivermectin is an antiparasitic drug that has shown also an effective pharmacological activity towards various infective agents, including viruses. This paper proposes an alternative mechanism of action for this drug that makes it capable of having an antiviral action, also against the novel coronavirus, in addition to the processes already reported in literature.
Ivermectin [mixture of 22, 23-dihydroavermectin B1a (80%) and 22, 23-dihydroavermectin B1b (20%)] (Fig. (Fig.1)1) is a macrocyclic lactone with a broad-spectrum antiparasitic pharmacological activity (Gonzalez Canga et al. 2008). It is the safest and most effective semi-synthetic derivative of the entire class of avermectins, discovered in 1975 by Professor Satoshi Ōmura as fermentation products of the actinomycete bacterium Streptomyces avermitilis (Crump 2017) (later reclassified in S. avermectinius (Takahashi et al. 2002)). Its main pharmacodynamics is to bind some channel proteins for chlorine controlled by glutamate, typical of specific classes of invertebrates, causing a greater permeability to this electrolyte: all this causes a hyperpolarization of the cell membrane, blocking inhibitory neurotransmission in neurons and myocytes, resulting in paralysis and death (Geary 2005). Commercialized since 1981, its low cost, its high efficacy and safety, and the marked tropism for helminths (therefore with an almost zero impact on the biochemistry of human beings) have led to its inclusion in the twenty-first World Health Organization's List of Essential Medicines (World Health Organization 2019).

Ivermectin is a versatile drug with unique characteristics, which make it interesting also for basic and applied research (in particular for drug repurposing): it seems to reveal an antibacterial (Lim et al. 2013; Ashraf et al. 2018), antiviral, and anticancer activity (Juarez et al. 2018; Intuyod et al. 2019), besides being potentially useful for the treatment of some chronic pathologies (Ashraf and Prichard 2016; Ventre et al. 2017), result of an action on a wide range of cellular targets.
Regarding its role as an antiviral agent, its efficacy has been demonstrated on several viruses, both in vitro and in vivo. Among the many mechanisms by which it performs its function, the most consolidated one sees ivermectin as an inhibitor of nuclear transport mediated by the importin α/β1 heterodimer, responsible for the translocation of various viral species proteins (HIV-1, SV40), indispensable for their replication (Wagstaff et al. 2011; Wagstaff et al. 2012). This inhibition appears to affect a considerable number of RNA viruses (Jans et al. 2019; Caly et al. 2012), such as Dengue Virus 1-4 (DENV) (Tay et al. 2013), West Nile Virus (WNV) (Yang et al. 2020), Venezuelan Equine Encephalitis Virus (VEEV) (Lundberg et al. 2013), and Influenza (Gotz et al. 2016). In addition, ivermectin has been shown to be effective against the Pseudorabies virus (PRV, with a DNA-based genome), both in vitro and in vivo (Lv et al. 2018), using the same mechanism. Caly et al. (Caly et al. 2020) have recently shown that the drug also inhibits the replication of the SARS-CoV-2 virus in vitro, however not clarifying how it occurs. Since the causative agent of COVID-19 is an RNA virus, it can be reasonably expected an interference with the same proteins and the same molecular processes described above.
However, ivermectin could prove to be a powerful antiviral, therefore also useful for a possible treatment of the new coronavirus associated syndrome, even from a new perspective. This could happen assuming its role as an ionophore agent, only hinted in the recent past but never fully described (Juarez et al. 2018). Ionophores are molecules that typically have a hydrophilic pocket which constitutes a specific binding site for one or more ions (usually cations), while its external surface is hydrophobic, allowing the complex thus formed to cross the cell membranes, affecting the hydro-electrolyte balance (Freedman 2012). These chemical species have historically been used to study the mitochondrial respiratory chain and ATP synthesis in eukaryotes (in this case also known as decoupling agents, such as 2, 4-dinitrophenol), and their antibiotic activity has long been appreciated (Bakker 1979). It is also hypothesized their role as antiviral drugs (Krenn et al. 2009; Sandler et al. 2020) and anticancer chemotherapeutic agents (Kaushik et al. 2018). Thinking of the structure of two of the most important ionophores, monensin A and valinomycin, respectively a polyether and a depsipeptide antibiotic, it is clear that they internally present many oxygen atoms (with related free electron doublets), indispensable for binding cations and transporting them through phospholipidic bilayers.
At a first glance, the two structures that make up the ivermectin formula do not have these chemical properties, nor those mentioned above, essential for a compound to be defined as ionophore. However, it can be hypothesized that two ivermectin molecules, reacting with each other in a “head-tail” mode, can create a complex suitable to be considered such (Fig. (Fig.2).2). This interaction could occur spontaneously or be mediated by the binding of the same molecules to some plasma transport proteins, in particular albumin (Klotz et al. 1990), which would have the role of positioning them in the correct way to obtain the proposed configuration.

As it can be seen, in this way, an internal cavity is formed: the oxygen atoms (indicated in red), now present in greater number, work as Lewis bases and could therefore coordinate a series of cations (Lewis acids). On the other hand, the –OH groups are highlighted in blue and they could have a decisive role in the stabilization of the new structure, with the establishment of chemical bonds between these functional groups: one or more –O– bridges (however, it is difficult the formation of ether bonds, since acid catalysis at high temperature is not possible under normal conditions, both in vitro and in vivo) or more probably hydrogen bonds could be formed, even among more molecular complexes of this type. However, the formation of other weak and strong interactions of various kinds cannot be excluded. Otherwise, specific cations could bind the two molecules in the proposed way, creating themselves the final structure and stabilizing it: there are examples already known in literature (Abbott et al. 1979). The external part of the complex, then, would already have in itself all the hydrophobic characteristics necessary to carry ions through the viral membrane. As a consequence, it would be determined an ionic imbalance between the external and internal environment, with the recall of water and consequent osmotic lysis. This would allow to neutralize the virus at an early stage of the infection, before; therefore, it can adhere to the host cells and enter it to exploit their biochemical machinery for the production of other viral particles. However, this hypothesis would concern only viruses without a proteic capsid, a structure that shows a certain resistance to osmotic pressure, even if to a lesser extent than a bacterial, fungal, or plant cell wall (Cordova et al. 2003). The new coronavirus is one of these, presenting only a phospholipid envelope in defense of the genetic material, where its few proteins are inserted and which it acquires in the act of exiting the infected cells (Sigrist et al. 2020). This unconventional electrolyte uptake mode could also affect the potential of the viral membrane, threatening its integrity and functionality. The same goes for the viral proteins present here. Furthermore, the concentration variation of some cations, thus determined, could inhibit some key enzymes in the viral replication, such as RNA-dependent RNA polymerases (RdRp) (te Velthuis et al. 2010), already used as pharmacological targets.
Another indication in favor of a possible ionophore role for ivermectin comes from the analysis of molecular similarity that can be carried out through the Drugbank database (www.drugbank.ca). By setting a minimum similarity threshold for ivermectin equal to 0.7, about 14 results are obtained. Among the various selected molecules, the majority of which have antiparasitic and antibiotic activity (already not only on the market but also in the study and experimentation phase), a compound that has high structural similarity is nystatin (score of 0.72), an antimycotic drug with an ionophoric activity at the plasma membrane level, where it forms channels (Yamasaki et al. 2011; Stillwell 2016; Rang 2015).
Immediately afterwards, with a slightly lower similarity, it can be find amphotericin B and natamycin, all pharmacological molecules of assured ionophoric activity (score of 0.71 and 0.706, respectively) (Stillwell 2016; Rang 2015; Ramos 1989; Ikehara et al. 1986).
In conclusion, pending computational simulations and chemical-physical laboratory analysis, this hypothesis could be applied to other known pharmacological molecules, in order to identify compounds with probable ionophore nature to be used in research and clinical practice.
Authors’ contributions
All research phases (idea, drafting of the paper, and proofreading) were conducted by the only author, ER.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abbott BJ, Fukuda DS, Dorman DE, Occolowitz JL, Debono M, Farhner L. Microbial transformation of A23187, a divalent cation ionophore antibiotic. Antimicrob Agents Chemother. 1979;16(6):808–812. doi: 10.1128/AAC.16.6.808. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Ashraf S, Prichard R. Ivermectin exhibits potent anti-mitotic activity. Vet Parasitol. 2016;226:1–4. doi: 10.1016/j.vetpar.2016.06.015. [PubMed] [CrossRef] [Google Scholar]
- Ashraf S, Chaudhry U, Raza A, Ghosh D, Zhao X. In vitro activity of ivermectin against Staphylococcus aureus clinical isolates. Antimicrob Resist Infect Control. 2018;7:27. doi: 10.1186/s13756-018-0314-4. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Bakker EP. Ionophore Antibiotics. In: Hahn F.E. (eds) Mechanism of action of antibacterial agents. Antibiotics. 1979;5:1. [Google Scholar]
- Caly L, Wagstaff KM, Jans DA. Nuclear trafficking of proteins from RNA viruses: potential target for anti-virals? Antiviral research. 2012;95:202–206. doi: 10.1016/j.antiviral.2012.06.008. [PubMed] [CrossRef] [Google Scholar]
- Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA-approved Drug Ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res. 2020;3:104787. doi: 10.1016/j.antiviral.2020.104787. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Cordova A, Deserno M, Gelbart WM, Ben-Shaul A. Osmotic shock and the strength of viral capsids. Biophys J. 2003;85(1):70–74. doi: 10.1016/S0006-3495(03)74455-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Crump A. Ivermectin: enigmatic multifaceted ‘wonder’ drug continues to surprise and exceed expectations. J Antibiot (Tokyo). 2017;70(5):495–505. doi: 10.1038/ja.2017.11. [PubMed] [CrossRef] [Google Scholar]
- Freedman JC (2012) Chapter 4 - Ionophores in planar lipid bilayers. In: Sperelakis N (ed) Cell Physiology Source Book, 4th edn. Academic Press, pp 61–66 ISBN 9780123877383
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug. Trends Parasitol. 2005;21(11):530–532. doi: 10.1016/j.pt.2005.08.014. [PubMed] [CrossRef] [Google Scholar]
- Gonzalez Canga A, et al. The pharmacokinetics and interactions of ivermectin in humans--a mini-review. AAPS J. 2008;10(1):42–46. doi: 10.1208/s12248-007-9000-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Gotz V, et al. Influenza A viruses escape from MxA restriction at the expense of efficient nuclear vRNP import. Sci Rep. 2016;6:23138. doi: 10.1038/srep23138. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Ikehara T, Yamaguchi H, Hosokawa K, Yonezu T, Miyamoto H. Effects of nystatin on intracellular contents and membrane transport of alkali cations, and cell volume in HeLa cells. J Membr Biol. 1986;90(3):231–240. doi: 10.1007/BF01870129. [PubMed] [CrossRef] [Google Scholar]
- Intuyod K, Hahnvajanawong C, Pinlaor P, Pinlaor S. Anti-parasitic drug ivermectin exhibits potent anticancer activity against gemcitabine-resistant cholangiocarcinoma in vitro. Anticancer Res. 2019;39(9):4837–4843. doi: 10.21873/anticanres.13669. [PubMed] [CrossRef] [Google Scholar]
- Jans DA, Martin AJ, Wagstaff KM. Inhibitors of nuclear transport. Curr Opin Cell Biol. 2019;58:50–60. doi: 10.1016/j.ceb.2019.01.001. [PubMed] [CrossRef] [Google Scholar]
- Juarez M, Schcolnik-Cabrera A, Dueñas-Gonzalez A. The multitargeted drug ivermectin: from an antiparasitic agent to a repositioned cancer drug. Am J Cancer Res. 2018;8(2):317–331. [PMC free article] [PubMed] [Google Scholar]
- Kaushik V, Yakisich JS, Kumar A, Azad N, Iyer AKV. Ionophores: Potential use as anticancer drugs and chemosensitizers. Cancers (Basel) 2018;10(10):pii: E360. doi: 10.3390/cancers10100360. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Klotz U, Ogbuokiri JE, Okonkwo PO. Ivermectin binds avidly to plasma proteins. Eur J Clin Pharmacol. 1990;39(6):607–608. doi: 10.1007/BF00316107. [PubMed] [CrossRef] [Google Scholar]
- Krenn BM, Gaudernak E, Holzer B, Lanke K, Van Kuppeveld FJ, Seipelt J. Antiviral activity of the zinc ionophores pyrithione and hinokitiol against picornavirus infections. J Virol. 2009;83(1):58–64. doi: 10.1128/JVI.01543-08. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lim LE, Vilchèze C, Ng C, Jacobs WR, Jr, Ramón-García S, Thompson CJ. Anthelmintic avermectins kill Mycobacterium tuberculosis, including multidrug-resistant clinical strains. Antimicrob Agents Chemother. 2013;57(2):1040–1046. doi: 10.1128/AAC.01696-12. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lundberg L, Pinkham C, Baer A, Amaya M, Narayanan A, Wagstaff KM, Jans DA, Kehn-Hall K. Nuclear import and export inhibitors alter capsid protein distribution in mammalian cells and reduce Venezuelan Equine Encephalitis Virus replication. Antiviral Res. 2013;100(3):662–672. doi: 10.1016/j.antiviral.2013.10.004. [PubMed] [CrossRef] [Google Scholar]
- Lv C, Liu W, Wang B, Dang R, Qiu L, Ren J, Yan C, Yang Z, Wang X. Ivermectin inhibits DNA polymerase UL42 of pseudorabies virus entrance into the nucleus and proliferation of the virus in vitro and vivo. Antiviral Res. 2018;159:55–62. doi: 10.1016/j.antiviral.2018.09.010. [PubMed] [CrossRef] [Google Scholar]
- Ramos H. Attias de Murciano A, Cohen BE, Bolard J. The polyene antibiotic amphotericin B acts as a Ca2+ ionophore in sterol-containing liposomes. Biochim Biophys Acta. 1989;982(2):303–306. doi: 10.1016/0005-2736(89)90069-2. [PubMed] [CrossRef] [Google Scholar]
- Rang PH. Rang and Dale’s pharmacology. In: Dale, Maureen M, Flower RJ, Rod J, editors. 1945-, Henderson, G. (Graeme) (Eighth ed.). [United Kingdom] 2015. [Google Scholar]
- Sandler ZJ, Vu MN, Menachery VD, Mounce BC (2020) Novel ionophores active against La Crosse virus identified through rapid antiviral screening. bioRxiv doi. 10.1101/2020.01.21.914929 [PMC free article] [PubMed]
- Sigrist CJ, Bridge A, Le Mercier P. A potential role for integrins in host cell entry by SARS-CoV-2. Antiviral Res. 2020;177:104759. doi: 10.1016/j.antiviral.2020.104759. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Stillwell W (2016) Chapter 19 - Membrane Transport. In: Stillwell W (ed) An introduction to biological membranes, 2nd edn. Elsevier, pp 423–451 ISBN 9780444637727
- Takahashi Y, Matsumoto A, Seino A, Ueno J, Iwai Y, Omura S. Streptomyces avermectinius sp. nov., an avermectin-producing strain. Int J Syst Evol Microbiol. 2002;52(Pt 6):2163–2168. [PubMed] [Google Scholar]
- Tay MY, et al. Nuclear localization of dengue virus (DENV) 1-4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor ivermectin. Antiviral Res. 2013;99(3):301–306. doi: 10.1016/j.antiviral.2013.06.002. [PubMed] [CrossRef] [Google Scholar]
- te Velthuis AJ, van den Worm SH, Sims AC, Baric RS, Snijder EJ, van Hemert MJ. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog. 2010;6(11):e1001176. doi: 10.1371/journal.ppat.1001176. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Ventre E, Rozières A, Lenief V, Albert F, Rossio P, Laoubi L, Dombrowicz D, Staels B, Ulmann L, Julia V, Vial E, Jomard A, Hacini-Rachinel F, Nicolas JF, Vocanson M. Topical ivermectin improves allergic skin inflammation. Allergy. 2017;72(8):1212–1221. doi: 10.1111/all.13118. [PubMed] [CrossRef] [Google Scholar]
- Wagstaff KM, Rawlinson SM, Hearps AC, Jans DA. An AlphaScreen(R)-based assay for high-throughput screening for specific inhibitors of nuclear import. Journal of biomolecular screening. 2011;16(2):192–200. doi: 10.1177/1087057110390360. [PubMed] [CrossRef] [Google Scholar]
- Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA. Ivermectin is a specific inhibitor of importin alpha/beta mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. The Biochemical journal. 2012;443(3):851–856. doi: 10.1042/BJ20120150. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- World Health Organization . World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization; 2019. [Google Scholar]
- Yamasaki M, Tamura N, Nakamura K, Sasaki N, Murakami M, Rajapakshage W, Kumara B, Tamura Y, Lim SY, Ohta H, Takiguchi M. Effects and mechanisms of action of polyene macrolide antibiotic nystatin on Babesia gibsoni in vitro. J Parasitol. 2011;97(6):1190–1192. doi: 10.1645/GE-2799.1. [PubMed] [CrossRef] [Google Scholar]
- Yang SNY, et al. The broad spectrum antiviral ivermectin targets the host nuclear transport importin α/β1 heterodimer. Antiviral Res. 2020;177:104760. doi: 10.1016/j.antiviral.2020.104760. [PubMed] [CrossRef] [Google Scholar]
https://www.sciencedirect.com/science/article/pii/S0167488911001145
ReviewRegulation of nucleocytoplasmic trafficking of viral proteins: An integral role in pathogenesis?☆
Research highlights
► Nucleocytoplasmic trafficking of viral proteins is central to viral infection. ► Posttranslational modification is a key means to regulate viral protein trafficking. ► Nuclear trafficking of viral proteins can be a target for development of anti-virals.
Abbreviations
Keywords
1. Introduction
The mammalian cell is a highly organised, dynamic structure that compartmentalises its many functions into organelles such as the nucleus, Golgi, and endoplasmic reticulum. The nucleus retains the genetic material for cell maintenance and replication, whereby efficient signal dependent targeting of cellular proteins into or out of the nucleus, mediated by the importin (IMP) superfamily of transporters (see Fig. 2; Section 2) is required for the cell to function. During infection by various viruses, specific viral-encoded gene products exploit the host cell nucleocytoplasmic trafficking machinery to enter and exit the nucleus as part of the strategy of the virus to evade the host immune response and replicate productively. Many of these viral proteins appear not only to possess targeting signals mediating high affinity interaction with the cellular nuclear transport factors, but also show precise regulation thereof by phosphorylation of these interactions by cellular/virally encoded kinases or other enzymes (see Section 3).
This review will focus in detail on viral proteins for which there is evidence of regulated nucleocytoplasmic trafficking in infected cells, including gene products from DNA viruses such as simian virus 40 (SV40) large tumour antigen (T-ag), human cytomegalovirus (HCMV) processivity factor ppUL44, and the human papilloma virus (HPV) E1 protein, as well as the phospho “P” protein from the negative stranded RNA Rabies virus (RV). The regulatory mechanisms and evidence for a physiologically important role in the viral infectious cycle will be discussed (Section 4), with the implication being that the regulation of viral protein nuclear import is crucial for many diverse viruses, thereby representing a potential target for the future development of anti-viral agents.
2. Nucleocytoplasmic transport
All transport into and out of the nucleus occurs through the nuclear pore complexes (NPCs), macromolecular structures (> 60 MDa) that span the double lipid bilayer of the nuclear envelope (NE) [1], [2], [3], [4]. There are approximately 2000 NPCs per “typical” vertebrate cell, depending on the stage of the cell cycle and the cell type. NPC structure is typified by 8-fold symmetry, being made up of multiple proteins called nucleoporins (Nups) [5], [6], [7], [8] which occur in multiples of eight [9]. With the exception of certain peripheral, asymmetric Nups, most Nups localise on both sides of a symmetry axis in the plane of the NE [2], [9], and can be grouped into several classes based on homology and functional similarity [10], including (i) transmembrane Nups (i.e. POM121 and Gp210 in vertebrates), which anchor the NPC within the NE and are bound by (ii) structural Nups (c. 50% of all Nups), which contribute to the overall architecture of the NPC and represent the scaffold linking the transmembrane Nups and (iii) FG-Nups (c. 33% of all Nups/50% of the NPC mass), which are distinguished by the fact that they contain multiple FxFG (single letter amino acid code, where x is any amino acid) or GLFG motifs separated by varying numbers of charged or polar amino acids [2], [11]. Fig. 1 shows the distribution of specific FG-Nups within the NPC, highlighting their position throughout the NPC. A number of studies indicate that FG-Nups are integral to bidirectional active transport through the NPC because of their ability to interact transiently with IMPs [9], [12], [13], [14], [15], [16].
 Fig. 1
Fig. 1Translocation through the NPC of proteins > 45 kDa is generally mediated by members of the IMP superfamily of nuclear transporters, which includes 6 α and c. 20 β forms in humans. IMPαs are adaptors that function as heterodimers with IMPβ1 [1], [17], [18], [19] in nuclear import, whilst IMPβs can mediate transport in either direction through the NPC, with those mediating nuclear export called exportins (EXPs). IMPs/EXPs recognise specific sequences, nuclear localisation sequences (NLSs) or nuclear export signals (NESs) respectively within the cargo protein with which they interact, with the monomeric guanine nucleotide binding protein/GTPase Ran a key additional factor (see below) modulating cargo binding [17], [20].
Monopartite basic NLSs, such as that from SV40 T-ag (PKKKRKV132) [21], [22] and HCMV ppUL44 (PNTKKQK431) [23] as well as bipartite NLSs, which comprise two clusters of basic residues such as the HPV E1 NLS (KRK85-/-KKVKRR125) [24], are generally recognised by the IMPα/IMPβ1 heterodimer. All IMPβs including IMPβ1, in contrast, are able to mediate import or export of their cargoes without the need for IMPα or other adaptors, although the NLS/NES sequences have not been defined in many cases. NESs recognised by EXP-1 (Crm1) [25], [26], [27] comprise 3–4 hydrophobic residues interspersed with 1 to 3 non-hydrophobic residues (L-x2-3-(L,I,M,F,M)-x2-3-L-x-(L,I,V) [17], [20]), the classic example being the NES from HIV-1 Rev (LPPLERLTL83) [28].
As indicated above, IMP-dependent passage through the NPC is effected by transient interactions of the IMPβs with FG-Nups; Nup358 is proposed to play a key role in assembly of the IMP–cargo complex [29], [30], with a gradient of increasing affinity postulated to facilitate the passage of IMP–cargo complexes from cytoplasmic to nucleoplasmic side of the NPC (see [31]). In the case of nuclear import, release at the nuclear face requires Ran in its activated GTP-bound form to bind to the IMPβ to dissociate the import complex (Fig. 2 left). Nuclear export is analogous, where the EXP, only when in complex with RanGTP, recognises a NES within a cargo and forms a trimeric export complex (EXP/RanGTP/NES-cargo) that is able to translocate through the NPC through transient interactions with FG-Nups such as Nup98 [32] and the non-FG-Nup Tpr ([33]) on the nuclear side, and Nup214 [34] on the cytoplasmic side (see Fig. 1), where the complex is dissociated via GTP hydrolysis by Ran of GTP to GDP, facilitated by RanGTPase-activating protein (RanGAP) (Fig. 2 right) and Ran binding protein 1 (RanBP1) and/or the RanBP1-like domains of Nup358 [30].
 Fig. 2
Fig. 2Many viral proteins utilise the host cell nucleocytoplasmic trafficking machinery (Fig. 2) to achieve efficient nuclear import and/or export in order to carry out particular roles in viral replication and pathogenesis, and/or modulate the host cell cycle or innate immune response (see below and Table 1). The next sections examine a number of different viral proteins by way of illustrating the diverse mechanisms regulating viral protein nuclear import/export.
Table 1. Selected examples of viral proteins where regulation of nucleocytoplasmic trafficking is implicated in viral pathogenesis.


ND, not determined.
Abbreviations: CBP, CREB-binding protein; Cdk, cyclin dependent kinase; CK2, protein kinase CK2; dsDNA-PK, double stranded DNA-dependent protein kinase; GSK3, glycogen synthase kinase 3; HIPK2, homeodomain-interacting protein kinase 2; LANA2, latency associated nuclear antigen 2; LMB, leptomycin B; PKA, protein kinase A; PKC, protein kinase C; PML, promyelocytic leukaemia protein; WT, wild type.
aSingle letter code used for sequences; known/potential kinase sites (in bold blue), acetylation sites (in bold purple and underlined) or ubiquitination sites (in bold orange and underlined) are highlighted, with amino acid position in the protein of interest shown in the superscript.
Source: Refs. [60], [61], [62], [63], [64], [65], [66], [67], [69], [70], [71], [72], [73], [74], [75], [76], [77], [80], [81], [82], [83], [94], [95], [96], [98], [99], [100], [101], [102], [103], [104], [105], [106], [110], [116], [119], [125].
3. Regulation of nuclear transport
General mechanisms by which nucleocytoplasmic trafficking can be regulated include modulation of the levels and distribution of IMPs/EXPs [35], [36] as well as the number and/or composition of NPCs [2], [11]. Fine-tuning of the localisation/transport of a single protein or group of proteins, however, requires more specific modification, generally of the protein cargo itself rather than of the transport machinery. The best understood mechanism of regulating nuclear transport is through phosphorylation near the NLS/NES modifying recognition by IMP/EXP [37], [38], but modifications such as acetylation, ubiquitinylation and sumoylation have also been described [39], [40], [41] to regulate nucleocytoplasmic trafficking of cellular proteins such as the tumour suppressors p110Rb and p53, Survivin, nuclear factor NF-κB, the phosphatase PTEN and the NF-κB essential modulator NEMO [42], [43], [44], [45], [46], [47], [48]. It is significant in this context that viral proteins are often highly posttranslationally modified (see Table 1), including through the action of cyclin-dependent kinases (Cdks), which can serve to effect cell cycle-dependent modulation of nucleocytoplasmic trafficking. A specific example is HPV E1, which will be examined in more detail in Section 4.2.
3.1. The cellular nuclear transport machinery as a viral target
The NPC and the Nups that constitute it are thought to be passive in nucleocytoplasmic transport in most situations. However, NPC composition and Nup conformation can have an influence on the transport of IMPs/EXPs as well as cargoes. Since IMPs/EXPs appear to have different affinities for the FG-Nups (see Section 2), the presence or absence of certain FG-Nups may favour one set of transport factors/cargo over another [13], [14], [15], [16].
Certain viral proteins are known to act directly or indirectly on the NPC and IMPs/EXPs [49] in order to act to alter host cell functions. An example with respect to the NPC is the 3C protease from the picornavirus Rhinovirus [50], which is thought to target Nups153, 214 and 358 for specific degradation in order to impair host cell nucleocytoplasmic transport (see Section 2 and Fig. 1), and thereby dampen anti-viral responses [50]; altered NPC structures have also been visualised in cells infected by the closely related poliovirus [51]. 2A protease from both Rhinovirus and poliovirus appears to act similarly to 3C in this respect [52], [53], [54], implying that the NPC is a key target of picornaviruses to disrupt host cell transport processes, and lead to “host cell shut down” to enable viral replication to proceed unchecked in the cytoplasm.
IMPs/EXPs can also be targets of viral proteins. Ebola virus VP24, for example, binds to and sequesters IMPα1 [55], [56], [57] in the cytoplasm, whilst IMPα2 is similarly sequestered by severe acute respiratory syndrome (SARS) coronavirus ORF6 [58]. In both cases, the IMPα is prevented from playing its normal role in mediating nuclear import of the STAT (signal transducer and activator of transcription) proteins in response to interferon (IFN), as part of the innate immune response (see [59]). Thus, it seems that various cytoplasmically replicating RNA viruses disrupt the cellular nuclear transport machinery in order to subvert the host cell transport processes necessary for the anti-viral response.
In the case of DNA viruses that replicate in the nucleus, however, efficient nuclear entry of many viral components is crucial for replication, so that disrupting the host cell nuclear import apparatus would not be a viable strategy to ensure efficient replication. The next section discusses the ways in which IMPs and cellular kinases can be subverted to enable efficient nuclear transport of gene products from DNA viruses that are required in the nucleus for replication.
3.2. Specific switches regulating IMP/EXP mediated trafficking
As indicated, the most common posttranslational modification known to regulate nuclear transport is phosphorylation. A number of viral proteins are known to require specific phosphorylation in different ways for efficient nuclear accumulation, including T-ag (see Section 4.1), HCMV ppUL44, chicken anaemia virus (CAV) VP3 and many others [23], [37], [38], [49]. Phosphorylation can regulate nuclear transport (see Fig. 3) by 1) directly modulating the affinity of an NLS/NES for its IMP/EXP; 2) facilitating masking or unmasking (intramolecular masking) of an NLS/NES within the protein carrying it; or 3) effecting the binding or release of an NLS/NES binding factor that is not an IMP/EXP (intermolecular masking) [4], [38].
 Fig. 3
Fig. 3Table 1 summarises the mechanisms of regulation of nuclear import/export for a number of viral proteins for which nucleocytoplasmic trafficking is known to be important for the infectious cycle, with Fig. 3 illustrating several specific examples. As can be seen from Table 1, phosphorylation is a key modulator of nuclear transport of viral proteins, but other modifications, such as acetylation and ubiquitinylation, can also modulate nuclear transport.
Phosphorylation-mediated modulation of NLS/NES access, resulting in either inhibition (intramolecular masking) or enhancement of transport (see Fig. 3 and Table 1), is the most common means to regulate nuclear transport efficiency. The human T-cell leukaemia virus type 2 (HTLV-2) Rex protein (see Fig. 3) is an example; in its pre-mature (p24) form, the N-terminal IMPβ-recognised NLS is masked [107], [108], [109], but upon phosphorylation of T164 by protein kinase CK1 (CK1)/glycogen synthase kinase 3 (GSK3) [107], S151/153 is subsequently phosphorylated by CK1 to produce the active p26 form of the protein with an accessible NLS [107], [108], [109]. In the case of Kaposi's sarcoma-associated herpes virus LANA2 (latency-associated nuclear antigen 2), phosphorylation at T564 by Akt is believed to promote a conformational change that inhibits Crm1 binding to the NES [118]. A similar mechanism appears to apply to CAV VP3 (see Fig. 3) through the T108 phosphorylation site [114], [115], although phosphorylation in this case appears to only occur in transformed and not normal cells, making the nuclear targeting module of VP3 an exciting possibility for tumour-cell specific nuclear targeting. In the case of the SV40 T-ag protein, protein kinase CK2 (CK2) phosphorylation at S111/112 increases the affinity of recognition of the NLS by IMPα/β1, thereby accelerating the nuclear import rate c. 50-fold; this can be further enhanced by phosphorylation of the double-stranded DNA-dependent protein kinase (dsDNA-PK) site S120, which facilitates phosphorylation at the CK2 site, as well as IMPα/β1 recognition/nuclear import [85], [86], [87], [88], [89], [90]. In analogous fashion, HCMV ppUL44 is phosphorylated at S413 by CK2 to enable higher affinity recognition of the NLS by IMPα/β1 and increased nuclear import (see Fig. 3; [23]), and a similar mechanism appears to apply to the Adenovirus E1a protein (see Fig. 3), where phosphorylation by Cdk1 at S89 enhances Crm1-mediated nuclear export [111].
Intermolecular masking occurs when a heterologous protein prevents IMP/EXP recognition of normally accessible NLS/NES sequences in a cargo protein. Inhibitor protein I-κB is an example of a very specific cytoplasmic retention factor which binds to the NLS of the transcription factor NF-κB p65 to prevent IMPα/β1 interaction and thereby inhibit nuclear import. Upon activation of signal transduction, e.g. cytokine production during an immune response, I-κB is phosphorylated and degraded to unmask the p65 NLS and enable nuclear import [126], [127]. An example of a more general cytoplasmic retention factor that affects nuclear import of a number of different NLS-containing proteins, including SV40 T-ag and HCMV ppUL44 [78] is BRCA1 associated protein 2 (BRAP2). Intermolecular masking of the T-ag NLS by BRAP2 is dependent on phosphorylation of T124 by Cdk1 adjacent to the NLS (see Fig. 3 and Table 1), whilst PKA/PKC mediated phosphorylation of T427 within the ppUL44 NLS similarly facilitates interaction with BRAP2 and cytoplasmic retention [78]. Cellular proteins such as p53 and p21cip [78], [128], [129], [130], [131], [132], [133] which possess NLSs and adjacent phosphorylation sites resembling those of SV40 T-ag and HCMV ppUL44 also appear to be able to be recognised by BRAP2 and inhibited in terms of nuclear import.
The subcellular distribution of viral proteins is able to be precisely regulated by specific cellular mechanisms; this can be seen as representing part of the host cell anti-viral response, but is also able to be exploited by the various viruses to enhance replication. For example, although the inhibition of nuclear import of SV40 T-ag or HCMV ppUL44, by BRAP2 leads to slowing/prevention of viral replication, this may also contribute to viral replication by delaying it until the optimal stage of the cell cycle or cellular signal transduction state, which is achieved by the phosphorylation control of BRAP2 interaction with SV40 T-ag/HCMV ppUL44. The following section describes several specific examples where a physiological role of regulated nucleocytoplasmic trafficking is implicated in viral pathogenesis and/or the viral replication cycle.
4. Selected examples of regulation of subcellular trafficking of viral proteins
4.1. SV40 T-ag and HCMV ppUL44: multiple mechanisms of regulation of nuclear import through protein–protein interactions
SV40 virus replication uniquely is dependent on a single protein – T-ag – whose roles include as an initiation factor for viral DNA replication, dysregulation of the cell cycle and blocking apoptosis [134], [135]. T-ag's three main functional domains are the J domain (a.a. 1–82) that binds to hsc70, the constitutively expressed homologue of heat shock protein hsp70 [136], [137], the LxCxE motif (residues 103–107) that confers binding to the retinoblastoma (Rb) family of proteins p110Rb, p107Rb and p130Rb2 [138], [139], and a bipartite carboxyl-terminal domain (a.a. 351–450 and 533–626) that binds to the tumour suppressor p53 [137], [138], as well as the CREB binding protein (CBP) and the functional homologues, p300 and p400, all of which have roles in cell growth and transformation [140], [141]. T-ag's functions in replication are nuclear, as are the functions of the various host cell target proteins of T-ag; consistent with this, T-ag possesses a highly efficient NLS [21], [22], [142]. Early work showed that T-ag was a phosphoprotein [143], [144], with several clusters of phosphorylation sites [145], [146], [147] shown to be phosphorylated in SV40 infected cells and critical for T-ag function/virus replication (see Table 1) [148]. These include the CK2 site (S111/112) [85], [87], [88], the Cdk1 (cdc2) site (T124) [86] and the less well characterised CK1/GSK3 site (S106) [91], all of which affect virus replication [93], [148] as shown in Table 1, which summarises the effect of mutations at these sites on SV40 T-ag nuclear transport as well as SV40 pathogenesis/replication.
HCMV DNA replication occurs within the nucleus of the infected cell, through a “rolling circle” mechanism [149] that requires at least 6 essential virally encoded gene products [150], [151] which include the DNA holoenzyme complex, which is made up of a catalytic subunit (pUL54), and the phosphoprotein and processivity factor ppUL44 [79]. The ppUL44 N-terminal region possesses the ability to bind dsDNA in the absence of ATP and clamp loaders, and through its ability to bind to pUL54, can link pUL54 to DNA and stimulate DNA polymerase activity [79]. The N-terminal region also possesses dimerisation activity [152], [153]. Early in infection, ppUL44 localises to the nucleus through a C-terminally localised NLS (PNTKKQK431) [23], that also appears to be responsible, for “piggy-back” nuclear import of other viral replication fork proteins such as pUL54 and the uracil DNA glycosylase pUL114 [154], [155], whereby the proteins may assemble in the cytoplasm on the ppUL44 dimer before nuclear import ([156], [157]; Fig. 4). Importantly, ppUL44 is a target for cellular and viral kinases during infection [23], [158], [159], with several phosphorylation sites, including CK2 (S413) and PKC (T427) sites, N-terminal (a.a. 410–424) to the NLS [23], [78] (see Table 1 and Fig. 3, Fig. 4). This constellation of phosphorylation sites N-terminally proximal to the NLS is closely comparable to that of SV40 T-ag (see above) [23].
 Fig. 4
Fig. 44.1.1. Positive and negative regulation of nuclear import through specific phosphorylation
Detailed analysis of the transport kinetics of bacterially expressed proteins microinjected into hepatoma cells indicated that the T-ag NLS alone (residues 126–132) conferred a much slower rate of import than the NLS together with the N-terminal flanking residues (a.a. 111–132) which contains the various phosphorylation sites mentioned above [88], [89]. Deletion/mutation of the CK2 site S111/112 to prevent phosphorylation decreased the import rate [88], whilst D112 substitution enhanced nuclear import [87], [160]; although S111 can function in its absence as a CK2 site, S112 is the main site of CK2 phosphorylation [87]. The mechanism of enhanced nuclear import through the CK2 site is through phosphorylation increasing the affinity of T-ag NLS recognition by the IMPα/β1 heterodimer [85]. Negative charge at S120, the dsDNA-PK site, apart from facilitating CK2 phosphorylation at S111/112, also enhances IMPα/β1 binding to the NLS [90]. That phosphorylation of S111/112 to enhance nuclear accumulation of T-ag is physiologically important in SV40 replication is indicated by the fact that viruses with mutations in the CK2 site (S112 and/or both S111/112) have markedly slower kinetics of DNA replication, and reduced viability (> 50%) [93], [148].
Significantly, HCMV ppUL44 processivity factor has an NLS comparable to that of SV40 T-ag, together with an adjacent CK2 site (see Table 1) that acts to increase the affinity of recognition by IMPα/β1 and nuclear transport efficiency [23]. Since ppUL44 contributes to nuclear accumulation of other HCMV gene products such as pUL54 and pUL114 involved in virus replication, the enhancement of ppUL44 nuclear import by CK2 would appear to be crucial to HCMV, with inhibition of CK2 potentially a viable future anti-viral approach to inhibit HCMV replication [23], [156], [157], [161].
That CK2 is exploited by SV40 and HCMV and possibly other viruses, to enhance nuclear localisation of proteins involved in their DNA replication can be understood in terms of CK2 being ubiquitously expressed and constitutively active [162]. Intriguingly, certain viruses have been shown to directly control CK2 localisation as well as up regulate its expression. During HSV-1 infection, for example, the ICP27 protein is known to recruit CK2 from the nucleus to the cytoplasm, resulting in a 3.5-fold increased CK2 activity by 6 h post infection that enhances cytoplasmic localisation of phosphorylated ICP27 and thereby facilitates its role in shuttling HSV mRNAs from the nucleus [163]. Analogously, CK2 appears to be recruited from subnuclear structures to regulate intranuclear transport of ribosomal RNA during Adenovirus infection [164]. The implication is that CK2 activity is integral to infection in the case of a number of viruses, with more examples of viruses using CK2 to modulate subcellular localisation likely to be identified in the near future.
In contrast to the effects of phosphorylation at S111/112/120, Cdk-phosphorylation or Asp substitution of T124 adjacent to the NLS inhibits T-ag nuclear import [78], [86]. The mechanism of inhibition of nuclear import is not through preventing IMPα/β1 recognition of the NLS, but rather through negative charge enhancing binding of the cytoplasmic retention factor BRAP2, first identified as a binding partner of BRCA1 in a yeast-2-hybrid screen [165]; negative charge at T124 appears to enhance specific binding of BRAP2 to SV40 T-ag, thereby inhibiting nuclear import [78].
Analogously, BRAP2 has also been shown to bind the HCMV processivity factor ppUL44, dependent on negative charge at T427 within the NLS (see Table 1 and Fig. 4) [78], making BRAP2 the first example of a cellular negative regulator of nuclear import (NRNI) that inhibits nuclear bound viral cargo in a phosphorylation-dependent manner. Although this has only been shown thus far for gene products from dsDNA viruses, it seems likely that this may apply to other viruses/viral gene products. The fact that BRAP2 may represent a general cellular defence mechanism to stem viral replication is an intriguing idea that warrants further investigation to examine its full potential as an anti-viral agent. It should not be ignored, however, that, as alluded to above, cytoplasmic retention of viral proteins until an optimal cell cycle/signal transduction state of the cell is attained is a strategy utilised by many viruses to facilitate rather than prevent virus production/infectivity etc. That virus replication is optimal at particular stages of the cell cycle has been shown for Hepatitis C, Epstein–Barr Virus (EBV), varicella zoster virus (VZV), Kaposi's sarcoma-associated herpes virus (KSHV), as well as HPV [166], [167], [168], [169], [170].
4.1.2. Inhibition of nuclear import through p110Rb
Unlike HCMV ppUL44, SV40 T-ag, as indicated above, is able to bind Rb family members through the Rb binding site (RbBS) [134], [135]. The CK1/GSK3 site (S106) within the RbBS has been shown to be critical for transformation and viral replication (see Table 1; [93], [148]), correlating with the fact that negative charge at this site inhibits nuclear transport [92] through modulation of binding of p110Rb, but not other Rb family members. Deletion or mutation of critical residues in the RbBS relieves inhibition of nuclear import for T-ag proteins carrying the RbBS, whilst cancer cells lacking functional p110Rb show no reduction in nuclear transport due to the RbBS. Based on fluorescence recovery after photobleaching (FRAP) experiments, the mechanism of inhibition appears to be through cytoplasmic retention of the Rb–T-ag complex [92].
Significantly, other DNA tumour viruses gene products such as adenovirus E1a [171], [172], [173], JC and Bk virus T-ag proteins [174], [175], [176], and pUL97 from HCMV [177] all possess RbBS's analogous to that of SV40 T-ag. Although the conventional view is that viral proteins target p110Rb to impair its role in the cell cycle [178], [179], it does not seem unreasonable to speculate that p110Rb in turn may act on a number of transforming viruses by modulating the nuclear import of diverse viral proteins.
4.2. HPV E1 protein: cell cycle phosphorylation controls levels of nuclear protein
HPV has a particular tropism for squamous mucosal or cutaneous epithelia [180], where infection can trigger hyperproliferation of epithelial keratinocytes and benign warts in the case of certain “lower risk” HPV genotypes (e.g. 6 and 11) [181], [182], or can lead to malignant cancer [183], [184] in the case of certain “high risk” HPV genotypes (e.g. 16 and 18) [181], [182], [184]. Most infections are latent, however, where the viral DNA persists in the host as low copy number extrachromosomal plasmids in the basal germinal stratum as a result of low-level expression of the viral genes [180].
E1 is a 70 kDa site-specific ATP-dependent DNA helicase essential for virus replication, which is highly conserved amongst all HPV types, and is essential for viral replication and amplification [185], [186], [187]. Together with HPV E2 protein, which increases its affinity for DNA [188], [189], [190], [191], [192], [193], [194], E1 is able to bind to a specific binding element in the viral origin to act to facilitate origin DNA unwinding, recruit the host cell DNA polymerase α-primase complex, and thereby initiate viral DNA synthesis [195], [196], [197], [198], [199], [200], [201]. E1 performs its role in the nuclear compartment, which it accesses through a bipartite NLS (HPV-11 E1 83KRK/-/S89/-/S93/-/KKVKRR125 [24]). Between the basic amino acids of the bipartite NLS is a potent NES (HPV-11 E1 97NVANAVESEIS107PRLDAIKL115 [84]) that confers rapid export out of the nucleus through Crm1 [24], [84]. Bovine papillomavirus (BPV) E1 is functionally homologous to HPV E1, being able to substitute for HPV E1 in replicating the HPV genome, and vice versa [185]. Although phosphorylation by Cdk2 at S-phase of the cell cycle promotes BPV E1 nuclear export via Crm1, where phosphorylation/dephosphorylation at S283 would appear to enable rapid nucleocytoplasmic shuttling [112], [113], Cdk phosphorylation appears to promote nuclear retention in the case of HPV-11 E1 [24], [84]. The presence of a NES in E1 presumably relates to the need for the virus to slow viral replication to establish a persistent infection in the basal keratinocytes and maintain low copy number by keeping the nuclear concentration of E1 low. Only once the basal cells differentiate and start to rise to the skin surface does E1 accumulate in the nucleus to enable HPV enter the vegetative stage of its life cycle, when the viral promoters are significantly up-regulated and late gene products produced to enable HPV to have the best chance to reinfect another host [170], [201], [202], [203], [204].
HPV E1 nuclear localisation is modulated by phosphorylation by MAPKs present in the cytoplasm, and Cdks in the nucleus, at specific stages of the cell cycle [24]. E1 has been shown to interact with several cyclin/Cdk complexes in vitro [68], [117], as well as directly with cyclin E [117], [205], an interaction that is essential for viral replication [117]. The N-terminal domain of E1 possesses a cyclin binding motif (RxL126) [117] as well as several Cdk phosphorylation sites, of which S89, S93 and S107 have been shown through mutational analysis to inhibit transient replication of viral origin-containing plasmids in transfected cells [117]. Phosphorylation of all three serines (see Fig. 5) appears to be required for efficient nuclear localisation that is dependent on active cyclin E/Cdk2 and/or cyclin A/Cdk2 at S107 [24], [84], [117], and MAPKs (ERK/JNK) at S89/93 [24]. S106 phosphorylation prevents Crm1 recognition of E1's NES [24], [84], whilst phosphorylation probably by MAPKs of S89 and S93 seems likely to facilitate recognition by IMPα/β1 [24], probably in a manner similar to the effect of the CK2 sites near/within the NLSs of T-ag and ppUL44 (see Section 4.1.1; Fig. 4, Fig. 5).
 Fig. 5
Fig. 5Phosphorylation of HPV E1 by Cdk has been shown to be crucial for viral replication; mutation of all four Cdk sites within the protein (including S107) impairs HPV replication in vitro and in vivo, without affecting association with HPV E2 or cyclin E [84], [117]. It would thus appear that precise regulation of nucleocytoplasmic shuttling of HPV E1 is necessary in order to modulate the levels of E1 nuclear activity according to the differentiation state of the host cell; premature nuclear entry of E1 leading to virus production before the cell reaches the skin surface would be counterproductive, and so cell cycle-dependent phosphorylation appears to be exploited to HPV's advantage to fine tune the levels of E1 in the nucleus in order to optimise its chance of infecting a new host.
4.3. Rabies virus P protein
Although RNA viruses replicate in the cytoplasm, specific gene products from many of them are known to enter the nucleus (see Table 1) and/or alter nuclear transport of key cellular factors directly or indirectly (see Section 3). Nuclear localising proteins from RNA viruses generally interfere with transcription factors involved in signalling related to the innate immune system, though direct binding, or indirect effects [59]. STATs are the key factors involved in the innate immune response targeted by many RNA viruses, including Nipah, Sendai, measles and RV [59], [206], [207].
RV, genus lyssavirus, family Rhabdoviridea is a neurotropical virus, possessing a small 12 kb, negative stranded RNA genome comprising only five genes [208], [209]. Through a leaky scanning translation mechanism, the gene encoding the RV phospho (P)-protein (RPP) produces 5 forms (P1–5, where P1 is the full length protein), which have been implicated in various important functions in the viral life cycle [209], [210]. These include as a cofactor in viral genome replication through binding of its N-terminal 19 amino acids (only present in the P1) to the RV polymerase (L), and as a chaperone for nucleoprotein (N) either through direct binding (through a.a. 1–177) or indirectly bound to viral RNA genome (N-RNA) through the C-terminal domain (CTD, a.a. 174–297, present in all forms of RPP). Importantly, however, RPP also plays a key role as an antagonist of the host anti-viral response in part though binding to nuclear factors such as the transcription factor STAT-1 and promyelocytic leukaemia tumour suppressor protein (PML) [211], [212], [213], [214], [215] also through the CTD. RPP is also able to interact in two distinct modes with the host cell microtubule (MT) system, either through a dynein light chain (DLC) associated sequence (DLC-AS; a.a. 139–151, present in all forms of RPP) which confers interaction with DLC8 to enable dynein-facilitated nuclear import of RPP, or through a second distinct MT-association sequence (MT-AS, absent from P1 and P2), in combination with the RPP self-association domain (a.a. 54–139), which mediates dimerisation and causes the retention of associated STAT-1 on MTs, independent of DLC8, thereby preventing STAT-1 nuclear import and dampening the host cell response to IFNs [121], [210], [216].
4.3.1. Distinct roles of RPP P1 and P3 forms in the cytoplasm and nucleus
The key forms of RPP appear to be P1 (1–297) and P3 (53–297), which function predominantly in the cytoplasm and nucleus respectively, with the strong N-terminal NES (NES1) the main basis of predominantly cytoplasmic localisation of P1, in contrast to P3 that lacks it and is predominantly nuclear [120]. P1's key role is in the cytoplasm, both as a cofactor for replication, and as a binding partner of STAT-1 to prevent its role in IFN signalling, P3, however, can exist in either the nucleus, where its predominant role is to inhibit STAT-1 DNA binding activity, or in the cytoplasm, where, in a dimerised form, it prevents STAT-1 nuclear access by binding it and associating with MTs (Fig. 6). Multiple PKC sites [217] throughout RPP provide additional levels of control, the best understood of which is the PKC site at S210, which appears to function as a switch to inhibit the NLS (a.a. KKYK214 -/- R260) within the CTD [120], [122] and expose a second NES (NES2: NFEQLKM232) normally buried within the CTD (see Fig. 6). Whether other PKC sites (e.g. S63, S64 and S162) within RPP modulate MT interaction and/or regulate dimerisation of the RPP to enable the MT-dependent retention of associated STAT-1, is unknown at this stage. What is clear, however, is that RPP dimerisation is critical for STAT-1 cytoplasmic retention, since deletion of the self-association domain abolishes P3 MT association, instead facilitating MT-enhanced nuclear import via the DLC-AS [210], [216]; a heterologous dimerisation domain functionally can substitute for the RPP self-association domain to restore STAT-1 cytoplasmic retention [210].
 Fig. 6
Fig. 64.3.2. Nucleocytoplasmic trafficking of forms of P protein contributes to pathogenicity through targeting STAT-1
That nucleocytoplasmic trafficking of RPP is critical to RV infection is implied by analysis of an attenuated non-lethal chicken embryo (CE) cell-adapted strain (Ni-CE) of the highly pathogenic Nishigahara (Ni) strain of RV. A chimeric CE(NiP) virus containing the Ni-P gene in the Ni-CE genetic background, is highly pathogenic, implying that RPP is a key virulence factor, and that mutations in the RPP are likely to be responsible for reduced pathogenicity of Ni-CE compared to Ni [123]. Intriguingly, 4 of a total of 7 amino acid substitutions in the Ni-CE strain, compared to that of Ni, are located within/near NES1 (see Section 4.3.1), correlating with the fact that the Ni-CE RPP P1 is more nuclear in infected cells than the Ni RPP, and thereby less able to prevent STAT-1 nuclear translocation in response to IFNα treatment. The implication is that the RPP NES1 plays a critical role in infection by specifically antagonising STAT-1 nuclear translocation to activate IFN-stimulated genes [124], [210], [218].
Similar mechanisms of regulating viral replication and immune evasion appear to be employed by Nipah virus which possesses 3 forms of the P protein, P (709 a.a.), V, and W proteins (456 and 450 a.a., respectively); all share the same N-terminal domain but vary in the C-terminal domain through frame shifting during translation [219]. The cytoplasmic and nuclear localisation of the gene products appears to be crucial to inhibit STAT-1 activities by directly interacting with STAT-1 and preventing its activation [207], [220]; W protein is found in the nucleus and V and P found in the cytoplasm [207], [221]. Nipah produces V and W in addition to the full length P protein to target STAT-1, with the varying forms acting in the cytoplasm and nucleus combining to effect inhibition of the innate immune response pathway. RV and Nipah and presumably other viruses thus utilise multiple forms of the same gene product to target STAT-1 in either the cytoplasm or nucleus to prevent the up-regulation of IFN-stimulated genes and thereby dampen the innate immune response. That STAT-1 is a target for many different viruses is known e.g. the V protein of measles [222], [223], [224] and rinderpest [225] viruses also binds STAT-1 to inhibit anti-viral responses. Clearly, perturbing the nucleocytoplasmic shuttling ability of the key viral proteins that sequester STAT-1 in nucleus/cytoplasm would represent an important step towards preventing viral evasion of the innate immune system, as would preventing interaction of STAT-1 with the viral proteins themselves.
5. Conclusions and future research
Precise regulation of the function of specific viral proteins is central to viral replication and pathogenesis, and as discussed here, nucleocytoplasmic trafficking plays a critical role in the case of many DNA and even RNA viruses (see Table 1). Phosphorylation appears to be the main mechanism by which viral protein nucleocytoplasmic trafficking is regulated during the virus life cycle, involving various cellular kinases as well as virally encoded kinases. The ubiquitously expressed CK2 enhances nuclear localisation of specific proteins involved in replication in the case of DNA tumour viruses such as SV40 and HCMV, whilst Cdks have been shown to play a crucial role in maintaining a persistent HPV infection in the skin through modulating E1 localisation; viruses that encode their own kinases such as VZV (ORF66, see Table 1) are less reliant on cellular kinases for control over subcellular localisation. Clearly, targeting the activity of kinases, such as CK2 and Cdk in order to perturb viral protein subcellular trafficking using specific inhibitors, represents a potential anti-viral strategy, although hampered by the obvious problem of effects on normal cellular functions of conventional kinase inhibitors. Screening for and/or developing compounds that block the nuclear transport of specific viral proteins though disrupting their interaction with IMP/EXPs seems an intriguing alternative, whereby a counter screening approach could be used to discard inhibitors of general host protein–IMP interaction, in order to identify inhibitors specific to IMP–viral protein interaction without affecting cellular proteins (Wagstaff et al., manuscript in preparation). Along similar lines, a unique approach would be to screen for compounds that stabilise or enhance the interaction with negative regulators of nuclear import such as BRAP2 with the SV40 T-ag or HCMV ppUL44 proteins or p110Rb with SV40 T-ag etc., as a means to inhibit viral protein nuclear import and thereby virus production.
In conclusion, based on the results summarised here (e.g. Table 1) and elsewhere, viral protein nucleocytoplasmic trafficking is central to viral infection/pathogenesis in many cases. Developing reagents directed specifically towards nuclear import/export of viral proteins rather than inhibitors of general transport looms as a fruitful avenue of research. In the face of the growing need for therapeutics to combat the consistently emerging lethal zoonotic viral threats to human health, such as SARS, Ebola and Nipah, as well as more familiar lethal pathogens, such as HIV and influenza, this avenue should probably be exploited in the near future with some urgency.
No comments:
Post a Comment
Note: Only a member of this blog may post a comment.